

A Conversation with Jack Scannell: The Intractable ‘Sociotechnical’ Problems of Drug Discovery and Development, Genetic Triumphalism and Predictive Validity


Jack is one of the sharpest thinkers on the pharmaceutical industry, with a talent for coining terms that then propagate among industry insiders and into wider tech circles. He coined Eroom’s Law in 2012, and has had a large impact by establishing predictive validity as the dominant variable in drug R&D productivity.
In this interview we discuss:
Whether the four original causes of Eroom’s Law still hold, and if we’ve seen a change in the trend.
Why feeding AI with data from poor biological models simply increases the number of wrong answers you can generate per second, and the counterintuitive result that marginally better models beat marginally worse ones by a surprising margin.
“Magic shotguns, not magic bullets”: genetic triumphalism, how the meaning of a drug target quietly shifted from a pharmacologically inferred receptor to a gene product, and why the industry stayed reductionist even where reductionism does not work.
Why value creation and value capture come apart in markets for R&D, why the winnings may accrue to whoever controls the remaining bottlenecks, and why “solving all disease” in a decade mistakes a sociotechnical problem for a technoscientific one.
Pablo Lubroth: What are you working on now, and the path that brought you here? From neuroscience to equity research to drug-discovery economics and now building a company.
Jack Scannell: I run an early-stage biotech called Etheros. We are developing small molecule enzyme mimetics with applications in neurodegeneration.
I co-founded Etheros having previously set up and run a niche consultancy focused on screening and disease model validity. After all, pretty much everything that fails in people, around 90% of projects, presumably worked pretty well in all the mice and cell lines in which it was tested beforehand. I got very interested in the gap between models and people as a major driver of R&D productivity. I got there through working in drug and biotech investments, looking at long-term R&D productivity trends, which were very depressing.
As scientists would tell you, we had become much more efficient at DNA sequencing, or protein structure analysis, or computational drug design, so the input efficiencies in drug R&D had seen great improvement. But R&D output efficiency had been heading in the opposite direction.
I was in equity research on and off from around 2005 until around 2019. I did a stint in tech-bio drug discovery during that time as well. I got into equity research from consulting: I was at the Boston Consulting Group doing work in drug and biotech companies. And before that I was a neuroscientist, a mixture of wet neuroscience and computational. I went to university first to be a medical doctor, then decided that neuroscience was probably more interesting. So that is roughly my career in reverse order.
In 2012 you coined Eroom’s Law, the observation that the number of new drugs approved per billion dollars of R&D has roughly halved every nine years since 1950, the inverse of Moore’s Law. Are the four causes you named then, better than the Beatles, the cautious regulator, throw money at it, and the basic research brute-force bias, still the right diagnosis?
If you go from 1950 to about 2010, those buckets are still reasonably good, although I might rename the cautious regulator problem, because it seems to blame the regulator specifically. I think the regulator probably reflects wider social expectations. Also, most people would not want us to go back to 1950, considering the way patients were treated as “experimental material.” But those four buckets are broadly correct.
The throw money at it tendency, after the golden years of drug R&D economics, a supra-normal returns business at least from the 1960s until returns started falling around 2000, has probably changed a bit. And on the basic research brute-force bias, I have done more work since that has shed light on it. That bucket is generally correct, but I would describe its contents a bit differently now.
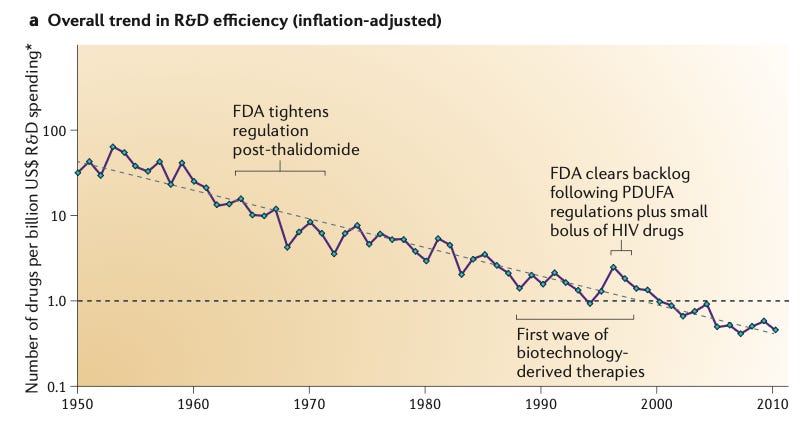
Better than the Beatles. Each new drug must beat an ever-improving back catalogue of mostly cheap generics that doctors and payers are content to keep using, so the bar for approval and reimbursement rises continuously and pushes R&D into harder diseases.
The cautious regulator. Regulatory risk tolerance has ratcheted downward since the 1962 Kefauver-Harris Amendment following thalidomide, and each drug scandal tightens requirements that rarely loosen, raising cost and lowering the odds of approval.
Throw money at it. The habit of responding to problems by adding money and people, which inflated R&D cost without a matching rise in approvals because returns were historically good and bigger budgets signalled status.
Basic research brute-force bias. The tendency to overestimate how much molecular biology and high-throughput screening of large compound libraries actually raise the odds a molecule proves safe and effective in humans, given that clinical success rates stayed flat for fifty years despite these tools.
Since 2012, what has improved, and what has gotten worse?
If we take a narrow definition from the first Eroom’s Law paper, the number of drugs per billion dollars spent fell exponentially from 1950 to 2010. Since then it has bumped around flat. So the trend is no longer negative, which is an improvement. But it has not started to go up.
If you look at other measures, like financial returns, they have a different historical trajectory, because R&D costs are one thing, but drug pricing and market growth are others. You had rather stable but very good supra-normal financial returns on R&D investment from as far back as I have data (the 1960s) to around 2000. That is for the industry in aggregate. Since 2000, financial returns on R&D have started to tail off, and in recent years, on fairly respectable measures, they were probably below the cost of capital. You can argue about precisely how low, and how you measure them, of course. Returns may have bumped up a little in the last year or two, mainly because the GLP-1s are skewing the economics of the whole industry.
So, in broad terms, most people would say things have stopped getting worse, but it is not obvious they have really gotten better.
Do agentic tooling, biological foundation models, and better physicochemical property prediction give you real reason to think Eroom’s Law can be bent?
There is no question that lots of those tools are incredibly useful. The real thing you have to think about, if you want to understand what they are going to do to overall R&D trends is whether they are a real step change or just an incremental change, and also why these improvements will have a net productivity hit that other huge improvements (DNA sequencing, x-ray crystallography, transgenic animal models, the creation of recombinant proteins, etc.) did not.
For lots of the quantitative tools you might now put under an AI rubric, there may have been substantial algorithmic evolution recently, but people have been using similar computational tools, earlier generations of them, for quite a long time. In many cases in biology, computational performance is data-limited rather than algorithm-limited. Not everything, but for some things. And the process to build new data is often quite slow.
Another thing, which is surprisingly absent in the AI productivity discussion, at least the parts of the discussion I see or read, is the extent to which AI changes the value that accrues to players in different parts of the R&D process. If something has become cheaper and more productive but other things remain bottlenecks, it may mean the people who control the bottlenecks make even more money than they used to, while the overall returns from the system as a whole change relatively little.
Does that mean that even more of the value will accrue to pharma and late stage development?
That may be true, and there are interesting economic precedents. I have done some work with a couple of game-theoretic economists which looked at where value accrues in markets for R&D. At least under certain model scenarios, if you are doing classic prisoner’s dilemma, Nash equilibrium type games in these markets, you find that the value created by certain technologies tends to accrue to consolidated downstream players. So there is a clear dissociation in markets for R&D between value creation and value capture.
Some of that VCs have learned by painful experience, not necessarily by game theoretic maths. There is the old joke that every platform company becomes a product company. That reflects the fact that it is hard to capture certain kinds of value. If AI substantially changes the costs and speed of some parts of the process, then you will have bottlenecks, or traffic jams, or oversupply elsewhere, and it is not obvious to me where the money goes.
The idea of your predictive validity work is already in this paper: the brute-force bias argues that better molecular tools may have manufactured the appearance of predictivity. If preclinical models inflate confidence in programs, what is the mechanism by which AI can fix that, rather than industrializing the same false confidence faster and cheaper?
Some of the first generation of tech-bio did not fix it at all, and simply reinvented the kind of biological assay problems the drug industry had first mis-industrialized years ago. Assays were optimized for easy, engineering-friendly data acquisition. They were not optimized for recapitulating important things relevant to humans with disease. I suspect a lot of what happened over the last 10 years hit that failure mode.
But some people think about it the right way. The predictive validity work says quality beats quantity, but the best thing is quality plus quantity. If you have quality and you can add quantity, that is very powerful. I feel enthusiastic about cases where people spend a lot of time and effort coming up with assays that capture the right biology and then find ways of increasing statistical performance or throughput via modern tools, some of which include AI.
Of course, I am generally looking from the outside so I am guessing what firms are doing to a large extent, but some start with a focus on human data and use AI tools to generate a volume and quality of information, and associated decision tools, in a way that could not have been done before. I guess that Noetik falls into this category, Axiom appears to be doing something cool in tox prediction. Then there are a few firms that I know a bit better, as an advisor: Orakl Oncology, Valid Therapeutics, and Biotx.ai who I think take the biology side of things extremely seriously as well as the AI tools.
So there are ways of doing the biology right and then getting more out of it with AI, and that seems a very sensible approach. But feeding AI with data from poor biological models simply increases the number of wrong answers you can generate per second.
Many concede that biology is more than one target and one mechanism, yet the industry stays addicted to molecular reductionism. What is the alternative in practice, and why has knowing the reductionist frame is wrong not been enough to displace it?
There is still a huge amount of what you might call genetic triumphalism, that molecular biology and genetics are the disciplines that have won biology. So it is not clear to me that everyone does think molecular reductionism is wrong. And in many cases it may be the right approach (e.g., monogenetic diseases, viral infections) even if it has been underwhelming in psychiatry or with a lot of complex age-related pathologies.
But this is a really interesting question and I have few thoughts which are not necessarily a coherent whole. My guess is that there are two parts of the story. One is biological, another is to do with how industry and science work.
On the biology side, it is clear that some very important drugs - a big chunk of psychopharmacology and perhaps some of the tyrosine kinase inhibitors in oncology - are “magic shotguns” and not magic bullets. In large parts of psychopharmacology, at least when I last looked, there was no easy relationship between the pattern of binding across lots of receptors and what the drugs did to people in a phenotypic sense. It is also interesting that genomics, when I last checked, has not yielded much. I worked at a tech-bio firm, nearly fifteen years ago, and one of the big ideas there was that we could model biological networks and then use chemoproteomic data to look at the “chords” that drugs were playing across the network.
Another thing is that drugs don’t know they are meant to have a target. They bind what they bind. When you apply more and better assays, you tend to find the same drug binds to more things. I think it is at least plausible that some kind of “cryptic polypharmacy” is more common and more therapeutically important than people think when drugs reach patients. So we shouldn’t confuse neat stories about targets, the creation myths, with how drugs actually work.
I think there is also an important industrial component. In the 1950s or 1960s, there were still some diseases where you could get a very good and quick phenotypic read-out in models or even in people, which meant you could sometimes run a design-make-test loop, or make serendipitous discoveries in people, without needing “the target”. This approach gave us drugs like metformin, or the first anti-psychotics and anti-depressants, all of which have messy or contested mechanisms even now.
But receptor theory had developed over the decades and in the 1960s, folks like James Black started having great success drugging receptors; think beta-blockers for angina. But it’s important to know that receptors, in the 1960s, were conceptually different to modern drug targets. A receptor was something whose existence you inferred on the basis of differential pharmacology. You would have a bunch of adrenergic agonists and antagonists, test them across a range of tissues, and on the basis of the differential response, infer that there were these things called adrenergic receptors, and that there were different classes of adrenergic receptors that did different things. But the physical or molecular identity of the receptor was not clear. So in the 60s and 70s, almost by definition, receptors as drug targets were pharmacologically tractable, because people used pharmacology to infer their existence. Also, assay read-outs would have looked phenotypic and functional by modern standards, I think it was a chunk of beating guinea pig heart for the beta blockers. This meant that the design-make-test loop automatically integrated across a bunch of different biological dimensions simultaneously.
By the 1980s and 1990s, the modern industrial view of drug targets had formed. They were single protein gene products. That realization, plus the boom in genomics, promised discovery factories with a more standard approach. You could kick off with some technology to find the most potent binders of your target (maybe high throughput screening) and apply a standard set of rules and cheap in vitro DMPK assays, but ignore biological sophistication until later. You get a set of “hits” that way, and plug them into a design-make-test loop to create “leads”, while keeping the target central to the process. Maybe monoclonal antibodies, with their super high specificity, also reinforced the importance of the drug target. Anyway, from an industrial perspective, this all looks much quicker, convenient and scalable than James Black with a lump of guinea pig heart.
But we know that R&D productivity declined spectacularly during this period. One problem in the early days of target-based drug discovery was that by first optimizing for potency, people created compounds that were really bad on lots of other dimensions that matter. This was then irrecoverable when the compounds made it into more biologically realistic assays. And the target-based approach also makes it impossible to discover drugs like metformin where there isn’t a clean target-based story.
What needs to occur for the dogma of molecular reductionism to change, and should it change?
My view is that if one can do drug discovery very close to people in ways that are not dangerous, or if one develops better human-derived models or good models in general, then maybe polypharmacology becomes a bit more tractable, or at least the target gets less important. But you still need a tractable design-make-test loop. You may not need single targets in that loop, but you need readouts that are interpretable, that are relatively quick, and which let you explore structure-activity relationships across the compounds you are making and testing.
I have talked about this kind of thing with Brian Warrington, a medicinal chemist who was a co-author on the 2012 Eroom’s Law paper. I think Brian is still busy discovering drugs today, but he worked with James Black on the H2 antagonists, back in the early 1970s. I recall that they did some optimisation using in vivo models, so they could measure stomach acid production (potency for this drug class), PK, and side effects all at the same time. Each design-make-test loop was slow and expensive and had very few compounds, but it was remarkably informative. It meant you didn’t get trapped in local optima, unlike the early days of target-based discovery where people first optimized only for potency.
I do not see in principle why one could not forget a bit about the single target and worry instead about assay performance across the relevant dimensions. Targets are not a logical necessity for the design-make-test loop. They are just a practical industrial implementation which might change if we could bring different kinds of assays to bear.
Does the regulatory preference and commercial requirement of having a molecular story preclude more drugs entering the clinic?
My view is that the FDA is probably not particularly wedded to targets and mechanisms. Drugs do still get approved when no one really knows how they work. Some drugs are approved with nice molecular stories about how they work that subsequently turn out to be wrong. During my academic life and subsequent professional career, the story about how lithium works has changed, and that does not stop lithium working. Another good example is anti-VEGF in oncology. When I first heard about them, people thought they starved tumors of vasculature which stopped them growing. Later, the story changed to the idea that they normalize tumor vasculature and hence chemotherapy drugs can get in.
Commercially, probably yes. And a simple story too. I have a friend who had to present his project to some important portfolio management committee at a mid-sized drug company. He had various slides with really complicated maps and models of biological pathways and, before his presentation, he was told to go away and come back with something much simpler, three or four boxes and a couple of arrows, “otherwise the board would never approve it”.
On the other hand, if you have human genetic causality, then we know that the target story is more likely to come true than some others. If you really don’t know what the target is, it puts you into the no-human-genetic-validation category, which is a disadvantage. That in turn means that you are basing your argument on data from model systems about which investors are likely to be sceptical. My view is that people could do a better job evaluating those model systems, but chances are that lots of venture capital firms and drug companies will not.
According to your work in predictive validity, a roughly 0.1 absolute improvement in the correlation between a model’s output and clinical outcome can offset a hundredfold increase in screening throughput. If that holds, most of the capital deployed in discovery over thirty years was spent on the wrong thing. Why has the field not reorganized around predictive validity, and is the answer that validity is almost unmeasurable until a program succeeds or fails, which makes the prescription hard to act on?
Generally yes, but let me disaggregate. I have never met anyone in the drug industry who thinks a bad model is better than a good model. But until the predictive-validity work and the decision-theoretic maths, which is not terribly complicated, most people probably did not understand the quantitative power of small changes in model validity. The non-obvious result is not that good models are better than bad models. It is that marginally better models are much better than marginally worse models. That is the surprising answer. Suddenly this thing that you already thought was important is more important than you previously thought. That result was not, and still is not, obvious to a lot of people.
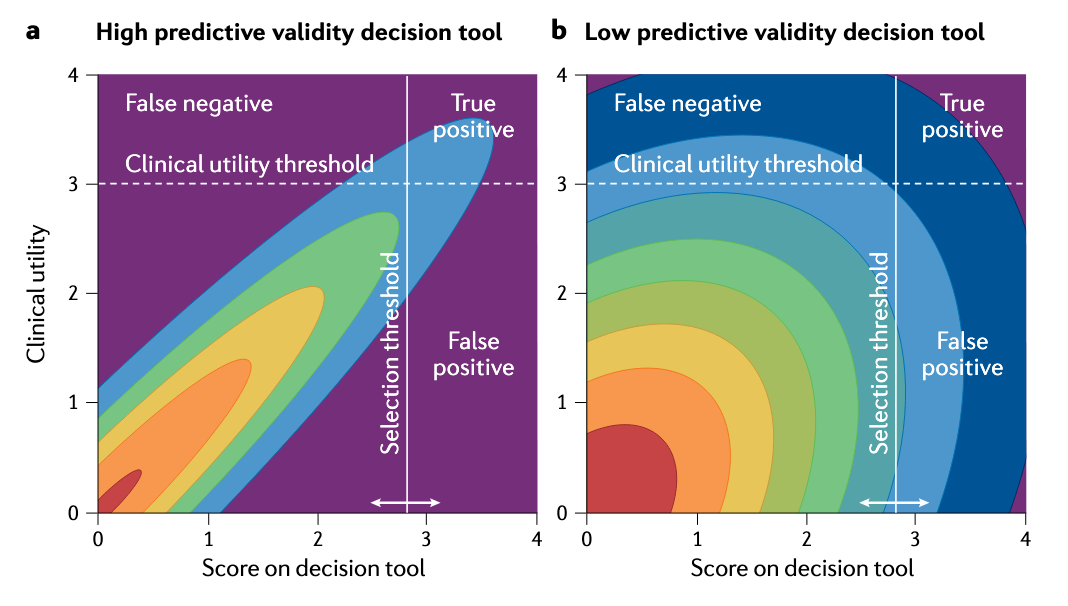
If you look at a lot of the R&D productivity literature, at least if you take a superficial view of it, it can make drug R&D look like a manufacturing process, where what really matters are things like cycle times and how many widgets we make and test. If we can make more widgets, we are going to get more stuff out of the production line. But that is not the real nature of the process. It is a decision process. People underestimated the quantitative power of better models for better decisions, and there is a superficial attraction to treating R&D as widget manufacturing.
Then you have real incentive problems. When I was doing the predictive validity work, I was not struck that I knew more about models than people in the drug industry. Every time I met people in the drug industry who knew about models, I realized they generally knew much more than I did. But their professional environment meant that they had to throw away the knowledge, or else other people running portfolios threw away that knowledge for them. Also, if you ask people working in a particular therapy area to evaluate the quality of their models, they will very rarely tell you they are rubbish, because they are worried that if they admit it, their programs get cut.
You have also got a problem around value capture. I talked to some sophisticated people working in oncology who said, look, we know the models we use are poor, but if we invested lots of money in making better models, the minute we publish phase 1b data and competitors learn what our target was, they can make antibodies or small molecules against that target without having invested in the model. So the economic value from better models is often hard to appropriate.
And if you look in academia, the most cynical statement is that an exciting false positive is much better for your career than a boring true negative. That is a cynical view, but people do not get Nature papers by doing long, tedious, gritty work to evaluate whether a particular mouse model of disease X recapitulates the human pathology and then responds the same way to drugs that have been in people. That is boring and industrial. It is not glamorous.
So you have a problem that the private sector struggles to appropriate the value of model-related innovation and for academics, it is not what they are paid to do. By the way, some drug companies know absolutely how important it is and do a very good job, but the overall quality is mixed and heterogeneous, and the incentives are tricky.
Is the AI revolution just another brute-force bias in the lineage of high-throughput screening, more compute and more data substituting for validity? What is different this time?
Again, it is mixed. I do come across AI led firms where the plan appears to be: we are going to implement some not particularly predictive biological model because it is the one that is easiest to industrialize into an AI-led design-make-test loop. The best that happens is you test more virtual in-silico ligands against a good in-silico model of an irrelevant cellular biological model. On the other hand, some people are doing it the other way around, thinking very hard about biology and then using the tools to allow models that are biologically realistic to cover more ground or do things they otherwise could not do.
How do you optimize for predictive validity inside your start-up, Etheros?
In a very unusual way, to be honest. I co-founded Etheros in 2022, and at the time my day job was thinking about predictive validity and disease models. I was consulting to biotech firms and drug companies. If you had asked me before Etheros came along whether I would be doing age-related neurodegeneration, I am almost certain I would have said no.
Etheros is developing small-molecule catalytic mimetics of superoxide dismutase, based on buckminsterfullerenes, to target superoxide, and tackling neurodegenerative conditions.
Etheros is an interesting case, because the data I came across, that motivated me to get involved, embodied predictive validity in a way that is different. Most of my thinking up to then had been divided into three buckets: First, how do you check whether the available models are good or bad predictors of human outcomes? Second, which human pathologies are unusually modelable, because those are the ones you should maybe work on? And third, where are there sudden increases in model quality, because those are places you might want to start deploying capital? Etheros did not fall into any of those categories.
I was put onto the Etheros science by Marc Feldmann, who pioneered the anti-TNFs, which are I think still the world’s best selling drug class in cumulative terms. Marc told me that he had come across something that looked like anti-TNF: an entirely new drug class; drugging a critical biological node that is conserved across species and in people; and which has a role in a large number of tissues and pathologies. Marc then introduced me to Laura Dugan, a neuroscientist based at Vanderbilt, who originally created some weird compounds that she thought of as chemical probes to help her dissect a particular part of biology associated with superoxide, or oxidative damage in neurons.
Over 25 years Laura had tested this technology in a remarkably diverse set of neural injury and neurodegeneration models, and all her work was placebo-controlled and double-blind. The species in which she tested them were mammals that diverged 100 million years ago. When I saw positive placebo-controlled, double-blind data from 12 different studies in macaques, rats, mice and pigs, where the neural injuries ranged from MPTP-induced parkinsonism, through spinal crush injury, through genetically induced ALS, through to normal aging, I realized that is a very unusual evidence structure. It is not one that people generally deliberately build very often when they are doing drug R&D.
Neurodegeneration models are typically wrong. But in Laura Dugan’s work I saw what I now jokingly call “uncorrelated wrongness”. The failure modes, the reasons why those models will fail to translate to people, are different from each other. It is a bit like having a set of unreliable and noisy signals: if the noise is uncorrelated and you add them together, what is left is signal. That is a rather different way of coming to what Marc Feldmann came to. It really does point to a critical node on diverse disease paths, because diverse upstream insults can be protected by blocking this node. It is a node that is probably conserved in people, because it is in primates and mice and rats and pigs. And it is probably relevant to heterogeneous, diverse human pathologies, because the upstream injuries across these models are so different to each other. There are fancy scientific names for this kind of triangulation, but it struck me as a very powerful signal that fitted into the predictive-validity framework. It is obvious in some of the more mathematical treatments, but I had not thought about it in practical terms until I came across Laura Dugan’s data. That is what got both me and Marc Feldmann excited. We thought Laura had this great technology that was neglected, so we dropped what we were doing to form a company to turn it into human medicine.
Walk us through the tripartite model from your 2025 work with Mellnik. How do you use it to judge the companies you assess in your investment work?
When John Mellnik and I were working on the tripartite model, I was consulting to a number of biotech companies that had platform technologies they thought would be better at discovering drugs in a particular domain, or measuring the performance of drugs in a particular domain, whether that was toxicology or some particular efficacy model.
The problem they found was that they would say, “we have got a better, more valid, way of measuring something important so we can make better decisions ”. But a common challenge was that the customers did not have very good ways of evaluating the claims, and nor did venture capital firms. And even if the customers or VCs did have ways of evaluating the truth claims, there was no way they could plug it into their financial models and calculate the value. This undermines incentives to invest in better models.
A question you could ask drug companies is: “suppose I have invented a much better way of doing phase 1b trials, so that you now have a much better idea about what is going to work downstream. How much would that be worth to you?” It turns out the DCF models they use have no way to answer that question. This makes no sense at all. Everyone knows R&D is a decision process, but it is modeled financially as if it is an attrition process. The financial models all use stage-specific costs and phase-transition parameters (e.g., the probability of going from preclinical to Phase 1, Phase 1 to Phase 2 etc.) and they treat the phase transitions as if they are statistically independent. That is, of course, nonsense. We do Phase 2 trials precisely because they are not statistically independent of Phase 3. If they were, there would be no point doing them. But all this means that standard drug and biotech valuation models are blind to decision quality. They contain no decision quality parameter that one can manipulate.
So to model decision quality and asset value as two interacting things, you disaggregate each of the steps in your modeling process. Rather than one probability of success at each step, you say the stepwise probability of success equals the probability the drug is good times the step’s true positive rate, plus the probability the drug is bad times the step’s false positive rate. If you disaggregate each step, then you can look at the logical downstream consequences of better upstream decisions, and you can say how much a better upstream decision is worth. You can parameterize these models, because if you know what the causes of failure are, you can start with FDA approvals and work back through each step of the portfolio. At each step, rather than having an indeterminate set of candidates that may be good or bad, you have an explicit mixture of things that are: (1) safe and effective; (2) safe and ineffective; (3) unsafe and effective; or (4) unsafe and ineffective. So you can work out the portfolio mix and decision quality and parameterize it, and work out how much that better phase 1b trial is worth, or how much you should invest in your decision tools rather than your assets, or how you should value an asset if the decision tools are better or worse than you expected.
How have I used this? Well, I used it in some work with Emulate, who make “organ on chips”. We used it to work out the financial value of better in vitro models of drug induced liver injury. I have also used it to help people argue with investors. Often people find that their company is being benchmarked on historical probability-of-success measures. If you can make reasonable arguments that the technology has got better, so that now in phase 1b you have some biomarkers that allow you to make a better decision, you can argue that the probability-of-technical-success assumptions being made are wrong. You might not be able to parameterize precisely how wrong they are, but you can make directional arguments. Or, if you can argue that for a given new modality, the a priori probability that this asset is good is higher than before, you find that it flows through the valuation process.
The idea of doing this was to give people tools so they were no longer blind to decision quality, in the hope that people would then spend a bit more money on making better decisions.
What is your most optimistic case for how AI improves discovery and development productivity, and whether it can bend Eroom’s Law? Do you think cycle times are compressible and trials shorter once you strip out human error and operational drag, or are those bound by biology and regulation in ways automation cannot reach?
I’ll start by saying that I feel as if I am a techno-optimist in general, but when I read what I write about R&D, I can look like a techno-pessimist. I do think that the wilder end of the optimistic prognoses about AI in drug R&D are just preposterous. But obviously it is really important and helpful in many respects.
If I was still an investment analyst, and not running a biotech, I would first separate out what you might call a narrow process or engineering view from a market-based view. For the process or engineering view, I would think about an idealized pipeline, apply changes to cycle times, cost, decision quality, etc., and come up with an answer. However, I don’t think that gets you far enough because the world has markets, not simply engineers. If you have very substantially changed the cost and timelines of certain steps in the R&D process, you change the structure of the market for R&D and that will trigger a bunch of responses.
A good example would be oncology R&D today. Cancer drug pricing in the US, and the value of cancer markets, has drawn in huge quantities of R&D capital. Arguably attrition rates have gone up because of that, because you have got so many candidates chasing a limited set of targets, and there are so few winners. I have not seen a proper analysis on this, but I would be very surprised if the average returns of the average player in US oncology R&D are high, because the size of the very obvious prize has created classic gold-rush economics. I suspect that if AI works as well as people hope, we are going to have a lot of gold-rush economics in certain parts of the supply chain, and the people doing the doing may not be the people who make the money.
Then there is another way of thinking about it, which I was put onto by a very good innovation scholar I know called Fred Steward, who has done a lot of work on energy transitions but also some serious work on drug R&D. He makes a distinction between what he calls a technoscientific problem and a sociotechnical problem, and I think this is a really useful way of thinking about the rise of AI in drug R&D and elsewhere.
A technoscientific innovation is like the classic Apollo moonshot: a problem where if you just set the engineers at it, and supply them with enough money, the problem will be solved. Sociotechnical innovation is very different. Sociotechnical innovation involves a complicated mix of economic, regulatory and process changes happening simultaneously over time, something like the building of modern healthcare systems, or the evolution of refrigeration in food supply and storage. My view is that AI in R&D is much more sociotechnical than technoscientific.
David Shaywitz has a lovely analogy here. If you look at the invention of electrical power in the US, it took a very long time for electrical power to transform the efficiency of American factories, despite the fact that they adopted electricity very quickly. Factories before electricity in the US would generally have a massive steam engine that drove a set of belts and shafts, which transmitted the power to the places where people worked. The design of the factory was constrained by the layout of the belts and shafts. When electricity first came along, they threw out the steam engine and put in one massive electric engine, but kept the belts and shafts the same. So electrical power first had a very small impact on productivity. Then, over the next 20 or 30 years, factories were rebuilt and reconfigured to use electrical power delivered to the workstations where people were. It was that reconfiguration of the factory which created huge productivity gains. I suspect that no one really knows what that reconfigured factory is going to look like when it comes to AI and drug R&D. I suspect many of the new biotech firms that call themselves AI-native are actually doing the AI equivalent of putting the electric engine into the old, steam-configured factory. The trajectory of complicated sociotechnical change is very difficult to call.
Thanks so much for such a fantastic conversation.