

BioByte 165: Proto as a New Programming Language, GPCR Biology May Explain Insulin Resistance, Bidirectional Fates for HSCs, Genome Editing with T Cells, and Alternate RNA Expands Proteome Diversity




Reminder to apply to attend our fourth annual AI x Bio Summit at the NYSE on July 23rd! Spots are filling up quickly, apply here to attend.

What we read
Papers
A high-level programming language for generative biology with Proto [Merchant et al., bioRxiv, June 2026]
Why it matters: Advances in generative methods for biology have rapidly improved our ability to design proteins and DNA sequences but researchers lack a general framework to integrate various tools for a desired design objective. Proto is a high-level programming language that decomposes design problems into modular workflows that can handle multiple objectives for tasks such as alternative intron splicing and promoter-repressor design.
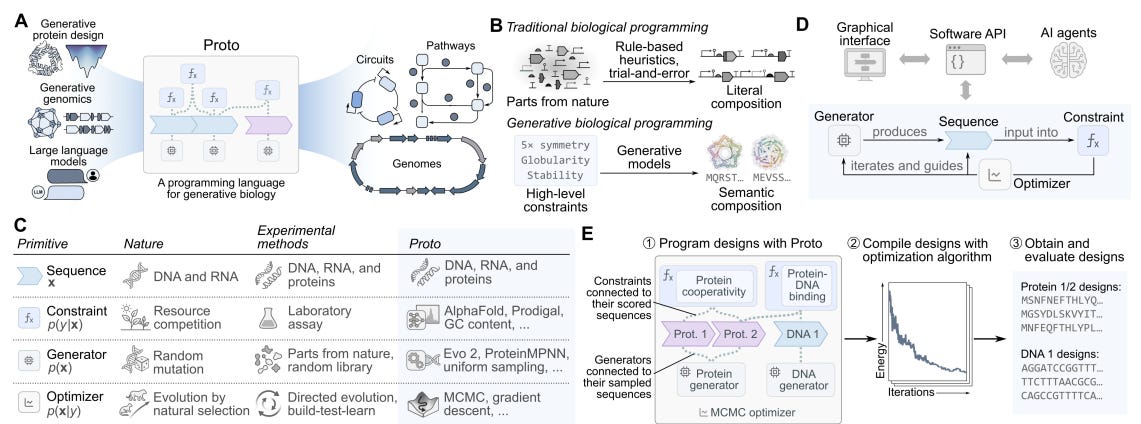
Despite the rapid advances of generative machine learning tools for biology, scientists looking to design proteins or genomic sequences must still rely on a heavy dose of intuition and domain expertise. Between protein and genomic design methods and large language models, researchers in theory now have access to all the tools one might need to execute build-test-learn cycles but are limited to the lack of a unifying design framework. To address this challenge, the authors of this paper developed Proto, a “high level programming language for generative biology” that aims to give scientists modular and semantic specifications of their work in a model agnostic manner.
Proto decomposes generative tasks in biology into four main components. Biomolecular sequences encoding proteins, RNA, and DNA are treated as a basic unit of input to a given design task. A sequence can be subjected to a “constraint” function that provides some notion of the sequence’s overall desirability (AlphaFold confidence metrics, FC content for DNA, etc.). New candidate sequences are provided by some “generator” function, which in the case of Proto can be a suite of protein and genomic sequence design tools such as autoregressive DNA language models, protein diffusion models, or even simple random mutation generators. Finally, sequences are refined and improved in an iterative manner by “optimizer” functions such as Markov Chain Monte Carlo (MCMC) and gradient descent methods based on the choice of generator function. By abstracting design tasks into these four modular components and bringing them to life with a user interface, the authors envisioned Proto as a tool ripe for “multi-objective, multi-modal, and multi-scale biological design.”
To demonstrate that Proto’s design language is useful, the authors began with an extensive reproduction effort across various domains. First, they showed that they could replicate previous symmetric protein homo-oligomer design studies using uniform mutation generators, ESMFold scoring as a constraint, and simulated ESMFold scoring as an optimizer. The team was also able to successfully replicate CRISPR-Cas design work using Evo1, chromatin accessibility sequence design with Evo2 and beam search, as well as antibody CDR design with gradient-based optimization for PD-L1 binders. Next, the team used Proto to rationally design genomic sequences with alternate splicing patterns across different human cell lines. AlphaGenome was integrated into Proto to use its cell-line-specific splice-site usage readout as a constraint, after which sequences were generated with a uniform mutation function and optimized with MCMC methods. Over 30% of new “ProtoIntron” sequences showed the specified alternate splicing patterns, compared to less than 7% of sequences from previous design efforts that tested nearly 1,000 sequences in total. Crucially, the authors emphasized that a combination of constraints were necessary for their success from a much smaller sequence pool, demonstrating the power of Proto’s capacity for multi-objective design specification. Finally, the team also used Proto to design new promoter-repressor pairs, testing the framework’s ability to generate successful sequences across protein and DNA modalities within the same experiment. ProtoPromoters were designed in two stages, first using Evo2 and rejection sampling constraints to find strong promoters, followed by uniform mutational proposal and MCMC optimization to add optimal palindromic operator sites and transcription start sites. ProtoRepressors were generated in a three stage manner by jointly optimizing over protein sequence, structural confidence, and promoter specificity using tools like AlphaFold2, LigandMPNN, and DeepPBS. Nearly half of generated promoters showed significant expression over no-promoter controls and 46% of repressor designs showed activity across 12 targets despite having very low sequence similarity to natural proteins. Importantly, this effort showed that such paired design objectives could be achieved without developing a new DNA-binder model and simply integrating existing, off-the-shelf tools.
Proto represents a significant engineering effort that goes beyond unifying various generative design tools by also providing a general framework to experiment with different constraints, generators, and optimizers as they become available. It will be interesting to see how the scientific community uses Proto’s API and user interface in conjunction with advances in agentic AI capabilities for various design tasks and if similar or alternate standards of specification arise.
Virion display reveals MD-1 as an endogenous agonist for the orphan receptor GPRC5B [Johansen et al., Science Signaling, June 2026]
Why it matters: Johansen et al. show how two high-throughput technologies can be combined to solve a major bottleneck in GPCR biology: identifying ligands for orphan receptors. By using this approach to deorphanize GPRC5B, they uncover an MD-1–GPRC5B macrophage–adipocyte signaling axis that may help explain obesity-linked adipose inflammation and insulin resistance.
Heng Zhu’s lab has been sprinting toward developing high-throughput technologies that enable characterization of the human proteome. Among many inventions, the lab builds on two previous in-house innovations. The first method tackles the challenge of purifying membrane proteins like GPCRs, which require lipid membranes to properly form and function. In 2013, the lab developed virion display, a parallelizable method for expressing membrane proteins in the lipid membranes of herpes simplex virus-1, validating that the GPCRs form proper conformations and retain function. The lab has also been gradually expanding technologies to display thousands of unique proteins on microarrays. In 2009, they expanded this to over 16,000 human proteins, creating the HuProt array and enabling comprehensive binding and activity screening against a large fraction of the human proteome. The technology has since been commercialized through CDI Labs.
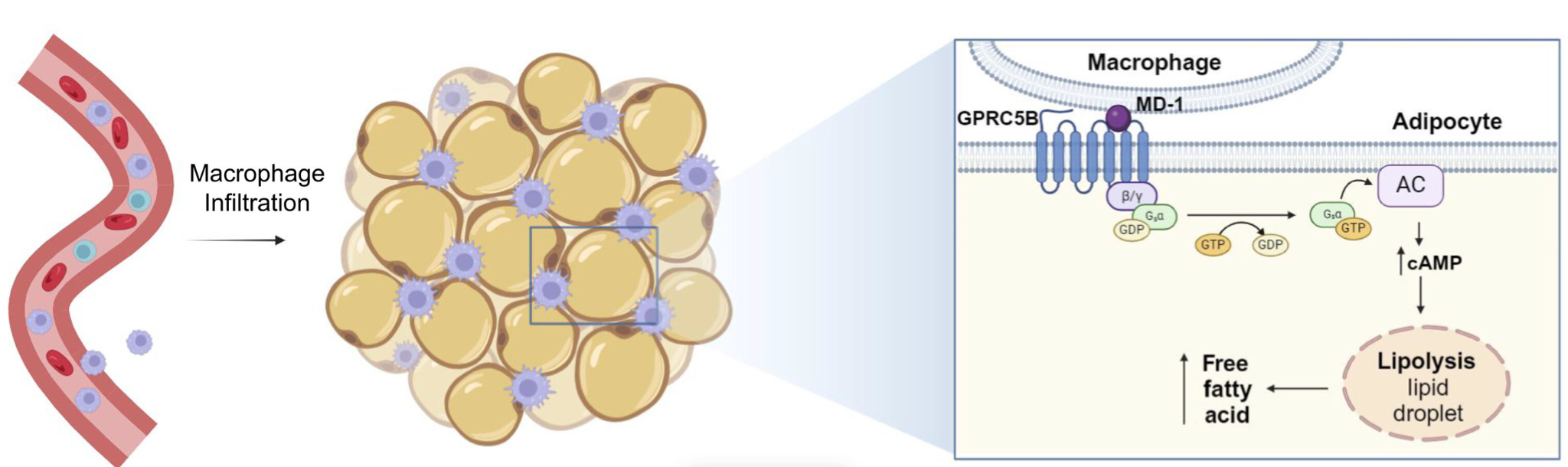
Johansen et al. build on these two pillars of technology to enable the deorphanization of a GPCR. What does this mean? One-third of nonodorant GPCRs are orphans - we don’t know what ligands activate them. Being able to identify which ligands GPCRs interact with would enable pathway-specific intervention and modulation that sit on a specific GPCR axis. Now let’s put two and two together. The Zhu lab developed a method to effectively display GPCRs embedded in a natural lipid membrane, enabling biochemical characterization and binding screening. Combine this with their HuProt array, and they’re able to screen against the human proteome to identify potential protein ligands for a GPCR of interest. They demonstrate this deorphanization capability against GPRC5B. A GPRC5B-KO mouse line is resistant to high-fat diet-induced obesity, adipose tissue inflammation, and insulin resistance - so GPRC5B may have a role in developing obesity through regulating adipose inflammation, but the specific ligands involved with GPRC5B are unknown.
To identify binders against GPRC5B, they flowed GPRC5B virions over the HuProt microarray and tagged where GPRC5B virions accumulated using an anti-HSV-1 antibody, anti-gD. Twenty-three human proteins accumulated with GPRC5B and formed the set of hypotheses. One of these caught the researchers’ eye: knockout of the MD-1 protein also leads to resistance to high-fat diet-induced obesity, adipose tissue inflammation, and insulin resistance - just like GPRC5B. They had a lead.
Now they needed to confirm a functional relationship between MD-1 and GPRC5B. They employed a battery of tests to identify the functional interaction. PRESTO-Tango confirmed that MD-1 agonism of GPRC5B led to β-arrestin recruitment. TRUPATpH BRET revealed that MD-1 agonism of GPRC5B drove signaling specifically through the G protein GαS.
Now to bring it back to the phenotype: does this agonism truly drive the adipose tissue inflammation and insulin resistance implicated in the mouse KO lines? To test this, they moved from biochemical interaction to a cell-based model of adipose tissue. Since GPRC5B is expressed on adipocytes and MD-1 is expressed by macrophages, they asked whether MD-1 could activate GPRC5B on adipocytes to induce lipolysis. In wild-type adipocytes, MD-1 increased release of nonesterified fatty acids, but this effect disappeared when GPRC5B was knocked down or knocked out. Then they brought macrophages into the system. Direct contact between macrophages and adipocytes drove a much stronger lipolytic response than soluble MD-1 alone, and this response required both macrophage MD-1 and adipocyte GPRC5B. This closes the loop: their technology-driven screen identified MD-1 as a candidate ligand, their signaling assays showed that MD-1 activates GPRC5B through GαS, and their coculture experiments connected that molecular interaction back to the obesity-linked phenotype by defining a macrophage-adipocyte signaling axis that regulates lipolysis.
Hematopoietic stem cells undergo bidirectional fate transitions in vivo [Fukushima et al., bioRxiv, June 2026]
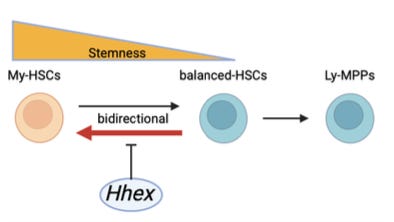
Why it matters: The standard model of blood renewal, or hematopoiesis, is a one-way hierarchy: hematopoietic stem cells (HSCs) sit upstream, then progressively shift toward less self-renewing, more lineage-committed states. Even within the pool of HSCs, there is an assumed unidirectionality. Myeloid-biased HSCs (My-HSCs) have often been treated as the higher-stemness (more primitive, self-renewing) state – cells that mainly produce myeloid blood lineages (red blood cells, platelets, granulocytes/monocytes), can give rise to more balanced HSCs (producing myeloid and lymphoid cells), and are not themselves thought to emerge from these balanced HSCs. Fukushima et al. challenge this model. Using clonal phylogenetic tracing in vivo, they demonstrate that lineage bias can move both ways – albeit asymmetrically: balanced HSCs frequently generate durable myeloid-biased HSCs, My-HSCs only rarely return toward balanced output, and the net dynamics favor progressive My-HSC accumulation over time. Age-associated myeloid skewing may reflect not just the expansion of fixed myeloid-biased clones, but the ongoing conversion of balanced clones into the self-renewing, myeloid-biased state.
The team built their case with a CP-tracer, a two-part clonal barcode delivered to HSCs by ex vivo lentiviral transduction before transplantation. The first part is a fixed, random DNA barcode marking which parent HSC a cell came from. The second is a ‘scratchpad’ – an array of CRISPR/Cas9 target sites that Cas9 cuts at random moments, leaving small heritable edits that accumulate as the founding HSC’s descendants divide. Since sibling cells inherit the edits present in their shared ancestor and add new ones, the pattern of shared versus private edits reconstructs the branching order within a clone – a phylogenetic tree. A single barcode can tell you a clone’s average output, but the scratchpad makes its internal history visible. This separates the CP-tracer from prior single-cell transplantation and marker-based studies that read only the aggregate output of a clone’s descendants (e.g. fate reversals surfaced, if at all, appeared as a shift in a clone’s average – not as a parent state visibly giving rise to another).
Across 181,695 descendant subclones from 847 parent cells, the authors recover sufficient lineage depth to reconstruct a phylogenetic tree that places balanced HSCs at a fork with two exits: one branch climbing back toward the self-renewing, higher-stemness My-HSC state, and the other descending toward more committed (differentiated) lymphoid-biased, multipotent progenitors. The ‘balanced to My-HSC’ route dominates, and the authors observe it in three ways. (1) Among HSC-repopulating subclones derived from balanced-HSC parents, 989 of 995 acquired My-HSC-like output, with lymphoid production suppressed or delayed. (2) 44 of 58 balanced-HSC parents each produced more than one myeloid-biased descendant. (3) Those My-HSC descendants stayed My-HSC through secondary transplantation – a durable fate switch. In tandem with this transition analysis, the tracing led the team to characterize two new functional groups in the stem-cell hierarchy – B-biased multipotent progenitors and transient myeloid progenitors.
Pairing the fate-tracing with scRNA-seq characterized the mechanistic layer. My-HSCs and balanced-HSCs do not resolve into separate clusters, but they carry distinct transcriptional programs, and RNA velocity points in the same balanced-to-myeloid direction. A clonal CRISPR screen of candidate regulators identified homeobox gene Hhex as the lead involved in the My-HSC / balanced-HSC bifurcation. Deleting Hhex increases My-HSCs and myeloid-restricted progenitors while reducing lymphoid-biased progenitors, and restoring Hhex rescues lineage bias in HSCs – but far less in downstream progenitors, suggesting that Hhex tilts the fate choice at the stem-cell stage. Multiomics analysis – RNA-seq, ATAC-seq, Hhex CUT&RUN – indicated that Hhex acts primarily by repressing the myeloid-biased program – closing regulatory sites at ETS/homeobox-motif-enriched regions – rather than by activating the lymphoid program. Hhex also declines in human bone-marrow HSCs with age, and its myeloid-suppressing effect is stronger in aged hematopoietic cells – an interesting link between the regulator and the myeloid drift of old blood.
The main caveat to the results follows from the method – the central tracing system depends on ex vivo transduction and transplantation as opposed to native, steady-state hematopoiesis. Reassuringly, however, the ‘balanced to My-HSC’ direction is supported by independent, non-transplant lineage datasets and human clonal hematopoiesis analyses. Cumulatively, HSC fate appears not to be a fixed ladder, but an asymmetric, partially reversible system of states whose myeloid-biased attractor strengthens with age.
Programming T cells for intercellular genome editing [Wasko et al., bioRxiv, June 2026]
Why it matters: Most delivery vehicles for therapeutic genome editing are passive: they circulate systemically, relying on diffusion and surface binding to find cells with clean markers to match the targeting logic built into the delivery shell. Getting the editor to the right tissue and cell population – without editing neighbors indiscriminately – remains a persistent issue. Wasko et al. develop an active approach: they engineer T-cells to sense a programmable ligand, and, only after that encounter, produce and transfer gene-editing cargo packaged in enveloped delivery vehicles (EDVs) – virus-inspired particles studded with a fusion protein. This circuit, termed Juxtacrine Enzyme Transfer (JET), shifts delivery from a particle-targeting problem to a programmable cell-cell behavior.
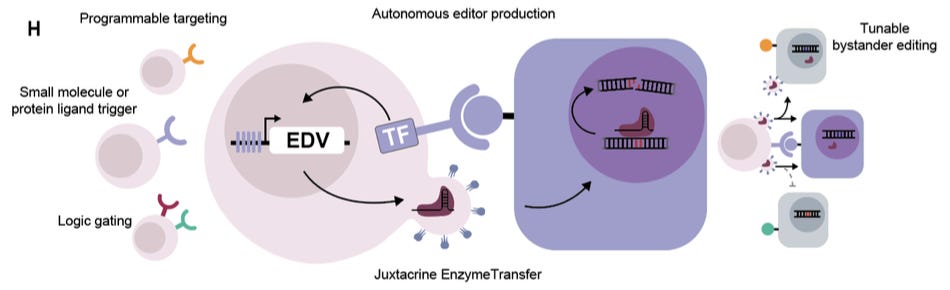
To demonstrate that a T-cell can transfer a functional editor, the team started in Jurkat cells (CD4+ T-cell line) carrying a doxycycline-inducible cassette encoding a miniGag-Cas9 fusion (a truncated HIV Gag protein that drives Cas9 into budding particles) and the fusogen VSV-G (helps cargo escape endosomes in the target cell). In the first test, the target cells (HEK293T) supplied the guide RNA (gRNA) against endogenous beta-2 microglobulin (B2M), a surface protein whose loss offers a clean flow-cytometry readout of successful Cas9 editing, so the T-cell only had to deliver Cas9 protein. A B2M loss of ~60% was recorded, a knockout requiring both doxycycline and the full miniGag/VSV-G-Cas9 system (cas9 or the tether without its fusogen edited nothing – ruling out leaky expression). The more difficult test had the Jurkats express both Cas9 and gRNA, so unmodified targets could lose B2M only if an intact, pre-assembled Cas9-guide complex was transferred. In this task, they achieved up to 18% B2M loss, proving feasibility.
The team next tuned the transfer architecture. VSV-G was swapped for Cocal-G, a related fusogen that resists inactivation by human serum – an eye toward eventual use in the bloodstream – with no loss of editing. More consequentially, replacing miniGag with an enveloped protein trimer (EPT) lets particles bud far more efficiently from Jurkats. Transwell and filtered-supernatant experiments justify EPT’s importance: when separated from their targets by a porous membrane, EPT producers behaved more like a secreted particle system and still edited well, while miniGag transfer largely required direct contact. With EPT, the T-cells delivered Cre recombinase – an enzyme that switches on a fluorescent reporter by excising a genomic stop cassette – into primary mouse neural progenitor cells and 3D gastrointestinal organoids, shifting the platform beyond flat HEK293T reporter assays.
Once the team established that the cells could make editors on command (via doxycycline), the focus shifted to target control. Here, production was gated with a synthetic Notch receptor (SNIPR): on contact with its antigen, the receptor is cleaved to release a transcription factor that switches on the EDV cassette, so the cell builds editors only where it meets its target. Against CD19, this transferred Cre into CD19+ cells while sparing CD19- controls – including NALM6 cells that carry CD19 at natural levels (e.g. endogenous), not just engineered overexpression. This same gate delivered Cas9 protein and Cas9 RNPs. From there, they illustrate more sophisticated programmability across three axes. (1) The modularity of the receptor was tested on six SNIPR-ligand pairs (CD19, EGFR, HER2, PSMA, LGR5, soluble TGF-beta), all firing only upon their respective matches. (2) The cells ran Boolean logic – an OR gate triggering on either of two antigens, an AND gate requiring a target antigen and doxycycline (antigen for place and drug for timing). (3) The scaffold choice defined the spatial spread: contact-biased miniGag confined editing to antigen-positive cells (even when rare), while diffusible EPT let editing reach neighboring cells – potentially useful when a target tissue has no clean marker of its own, but sits among cells that do. Finally, as an encouraging sign – the circuit ran in primary human T cells from three donors and performed CD19-specific Cre transfer with minimal target-cell cytotoxicity, unlike CAR-based methods.
The next stage is the move from ex vivo to in vivo: homing, persistence, immunogenicity data, and therapeutic editing efficacy are critical gates. The authors acknowledge current engineering obstacles – the circuit is large and integrates inefficiently into primary T-cells, resting T-cells may be too quiescent to produce particles without activation, and the VSV-G on the producer’s surface may invite immune clearance (though that may also be useful for capping editor output once the target is reached). Despite this, imbuing immune cells with the ability to seek and deliver an editor – codifying targeting as a property of programmable cell logic rather than of a particle’s surface – lays the groundwork for an exciting approach to this unrelenting challenge.
Alternate RNA decoding results in stable and abundant proteins in mammals [Tsour et. al., Nature, June 2026]
Why it matters: The central dogma assumes that protein diversity arises mostly from genetic variation, alternative splicing, and post-translational modifications, while translation faithfully decodes the genetic code. Although translational errors do occur, they’re considered rare and biologically inconsequential. This paper shows that alternate RNA coding is widespread in mammals, which generates abundant, stable, and tissue-specific protein variants that substantially expand proteomic diversity beyond the genome.
Tsour et al. integrate deep proteomic, transcriptomic, and genomic data from 1,094 human samples spanning 26 healthy tissues and 6 cancer types to systematically identify amino acid substitutions arising from translation rather than DNA mutations or RNA editing. After extensive validation, they identified 8,746 unique substituted peptides across 1,767 genes, demonstrating that alternate decoding is pervasive across mammalian proteomes. While many substitutions occur at low frequency, hundreds of proteins accumulate substituted proteoforms at levels comparable to – or even exceeding – their canonical counterparts, indicating that alternate decoding can become a dominant source of protein diversity.
The authors then investigate why some alternatively translated proteins become highly abundant. Substitution frequency depends on multiple features of the translation process, including codon usage, codon-anticodon mismatches, and RNA modifications. However, translation alone does not explain protein abundance. Using metabolic labeling experiments, they show that many substituted proteoforms exhibit substantially slower degradation than their canonical counterparts, allowing rare decoding events to accumulate into abundant protein populations.
Alternate decoding is also highly structured. Specific substitution types, tissues, and protein families exhibit reproducible substitution patterns across individuals. Highly abundant recoded proteins are enriched in transcription factors, signaling proteins, proteasome components, and RNA-processing factors, while certain substitutions are preferentially enriched in tumors relative to matched normal tissues. The authors further identify conserved substituted proteoforms in both humans and mice, which suggests that alternate RNA decoding is an evolutionarily conserved feature of mammalian translation.
This work reframes translation as a probabilistic decoding process that systematically expands the mammalian proteome. Rather than representing isolated translational errors, alternate decoding generates stable, abundant, and biologically regulated proteoforms, introducing an additional layer of proteome diversification beyond the genome and transcriptome.
Notable deals
Nura Bio raises $73.8M for their Series B in a round led by The Column Group. The proceeds of the raise will go towards the clinical development of their two lead programs, both of which are small molecule inhibitors of SARM1—a nicotinamide adenine dinucleotide hydrolase with a well-established role in axon degradation. Their lead asset, NB-4746, is currently enrolling for a combined Phase Ib/IIa for ALS, having already demonstrated favorable safety profiles in an earlier Phase I study. The following asset in their pipeline (NB-9402) is a covalent, allosteric small molecule which is currently in a Phase Ia study. This study is expected to conclude sometime in 2026. Other investors in the round included Euclidean Capital, Samsara BioCapital, and Sanofi Ventures.
London-based RQ Bio closes a $115M Series A. RQ seeks to address the seasonal variability of influenza via long-acting monoclonal antibodies which target epitopes that are less susceptible to seasonal variation. The company’s lead asset—RQB01—is currently in IND-enabling studies, leveraging this approach to help prevent serious disease in vulnerable patient populations. Frazier Life Sciences led the round, with participation from EQT Life Sciences, Forbion, LifeArc Ventures (their founding investor), Monograph, Oxford Science Enterprises, the University of Oxford, and Wellington Management.
In an oversubscribed Series B, Ollin Biosciences raises $330M to enable global Phase III trials of their lead asset, OLN324. The drug is a best-in-class bispecific antibody targeting VEGF and Ang2 which are known drivers of retinal vascular diseases. It will enter Phase III trials in the latter half of 2026 for both diabetic macular edema (DME) and wet age-related macular degeneration (wAMD), having already demonstrated superior therapeutic effect relative to the approved therapy faricimab (Vabysmo®). Funds will also be used for OLN102, a first-in-class bispecific antibody targeting TSHR and IGF-1R for thyroid eye disease. It is slated to enter clinical development sometime in 2026. OLN324 and OLN102 were discovered by and are being co-developed with Innovent Biologics and VelaVigo respectively. TCGX and ARCH Venture Partners led the round while a16z Bio+Health, Blackstone Multi-Asset Investing, Commodore Capital, Canada Pension Plan Investment Board (CPP Investments), Monograph Capital, Mubadala Capital, RA Capital Management, accounts advised by T. Rowe Price Investment Management, and a sovereign wealth fund also participated.
Osanni Bio announces the close of their $190M Series B led by Patient Square Capital. Funding will be used to advance their existing pipeline and further scale their developmental capabilities. Once the assets in their pipeline are sufficiently developed, they are passed off to one of Osanni’s affiliate companies (affectionately called Avians) which are more well-equipped to scale and commercialize. They currently have seven programs in the pipeline across ophthalmology, cardiology and cancer, with their lead for dry age-related macular degeneration finishing a Phase Ib trial outside the US. The Horowitz Group, Invus Opportunities, and the Retinal Degeneration Fund also participated in the round.

In case you missed it
Uneven Frontiers: How AI will transform biopharma—and why the sequence of change matters
What we liked on socials channels



Events

An event not to be missed: come to our flagship AI x Bio Summit! Hosted at the New York Stock Exchange on July 23, 2026. Talk to leaders in AI x Bio from firms like Anthropic, Enveda, Formation Bio, and on. Apply here → https://events.decodingbio.com/summit2026
Field Trip
Did we miss anything? Would you like to contribute to Decoding Bio by writing a guest post? Drop us a note here or chat with us on Twitter: @decodingbio.