

BioByte 161: CellCage Enclosures Allow Monitoring of Dynamic Transitions Across Cell States, New Breakthroughs in Targeting GPCRs, and a Modular View of Aging




Welcome to Decoding Bio’s BioByte: each week our writing collective highlight notable news—from the latest scientific papers to the latest funding rounds—and everything in between. All in one place.


What we read
Papers
Scalable longitudinal imaging and transcriptomics of cells in dynamic enclosures [Khurana et al., bioRxiv, May 2026]
Why it matters: Single-cell transcriptomics defines cell states by clustering RNA, but a sequencing readout is a static snapshot that destroys the cell and erases its history. Subsequently, scientists are left to label cells by what they express and infer their function, as opposed to observing what the cell does and investigating which genes drive it. The team from Cellanome circumvents this with a platform built on ‘CellCage Enclosures’ (CCEs): computer vision locates every cell in a flow-cell lane, and a digital micromirror device projects 405nm light to photopolymerize a hollow, porous hydrogel box around each one – tens of thousands per experiment (a lane in under 15 minutes). Caged cells are observed and imaged for days, then lysed in place so that ~23,000 surface capture spots per lane tie each cell’s endpoint transcriptome to its enclosure. The functional readout isn’t just morphology: surface proteins, secreted cytokines, and transcriptomes are logged per enclosure – effectively pairing a cell’s history with its molecular program.
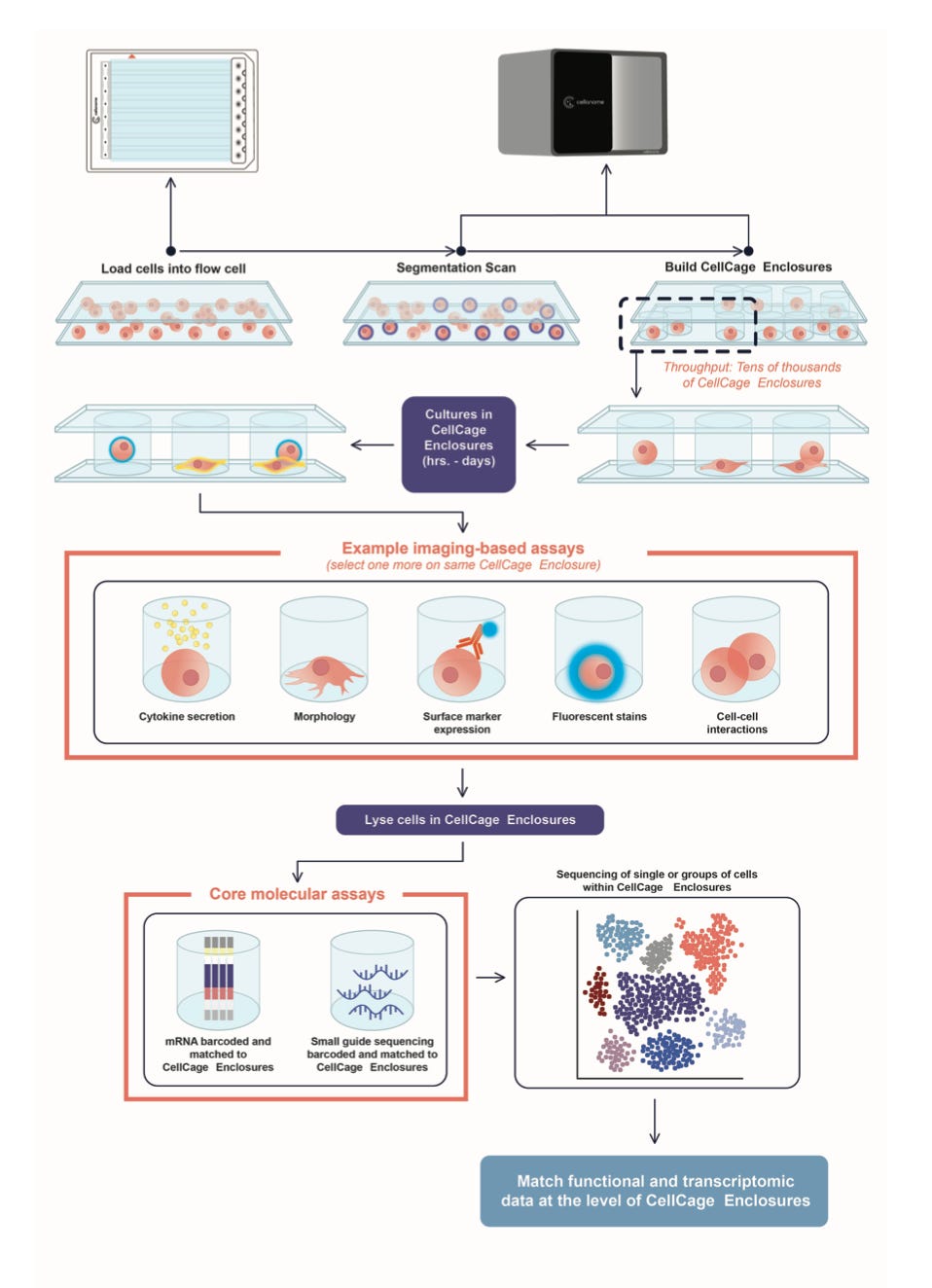
The recurring finding is that the genes controlling a function are not the genes that typically define transcriptomic clusters. Let’s look at the context of cancer drug resistance as an example. After imaging A549 lung cancer cells under the EGFR (epidermal growth factor receptor) inhibitor Olmutinib every 8 hours for 32 hours and then sequencing each enclosure, the team sorted ~10,600 treated enclosures into four behavioral phenotypes – including a ‘daughter-cell-resistant’ state, where a cell divides under drug pressure and one daughter dies while its sibling persists. These behaviors did not map onto transcriptomic clusters – every cluster held every phenotype. However, fusing the imaging-defined behaviors with gene expression clusters surfaced 15 enriched pathways invisible to either alone. The persister state carried upregulated calcium-activated potassium channels (KCNMA1 and KCNN4, the latter linked to chemoresistance) and a p53-dependent quiescence program (‘arrest genes’ holding cells in a dormant, slow-cycling state) in these p53-wild-type cells, implying a non-genetic drug tolerance in which siblings diverge in fate with no new mutation.
The team next evaluated the question more directly: can transcriptomics predict what a cell does? Two functions read out by fluorescence as ground truth – lipid loading in differentiating preadipocytes and particle engulfment (phagocytosis) by microglia – exhibited the same blind spot. Clustering failed to flag cells that were actually accumulating lipids and the most phagocytic, while models trained on the paired data recovered genuine drivers of each function – including phagocytosis genes whose expression barely varied across clusters and textbook complement proteins (which normally tag targets for engulfment) that turned out to be inversely related to phagocytic activity.
Those drivers emerged from natural cell-to-cell variability – the paired readout alone surfaced them, with no engineered perturbation. Doing this at scale with fickle cell types is what makes this compelling: forming dissolvable compartments on demand preserves its cage-adherent, large, and fragile cells (e.g. neurons) in a near-native state, something wells and droplet methods handle poorly. The main caveats to consider with this early progress include the discoveries being associations – not perturbationally proven mechanisms – and the expression-to-phenotype classifier topping out at 68% accuracy. However, encouraging strides are already being made – a companion preprint with Genentech applies the same enclosures to pooled CRISPR perturbation screens and adds proteomic readouts – toward the perturbation-rich, multi-cell, time-resolved regime that interaction studies and emerging virtual-cell models will need.
Taking cell therapy inside the body

Beyond traditional limits: explore the drug delivery technology opening new doors in genomic medicine.
This post is sponsored by Cytiva.
De novo design of miniproteins targeting GPCRs [Muratspahić et al., Nature, May 2026]
Why this matters: GPCRs are one of the most important drug target families, but they remain difficult to modulate because their activity depends on subtle conformational changes and they are membrane-embedded receptors. Muratspahić et al. combine computational protein design with high-throughput cellular screening to create de novo miniprotein modulators across diverse GPCRs, including new agonists and antagonists.
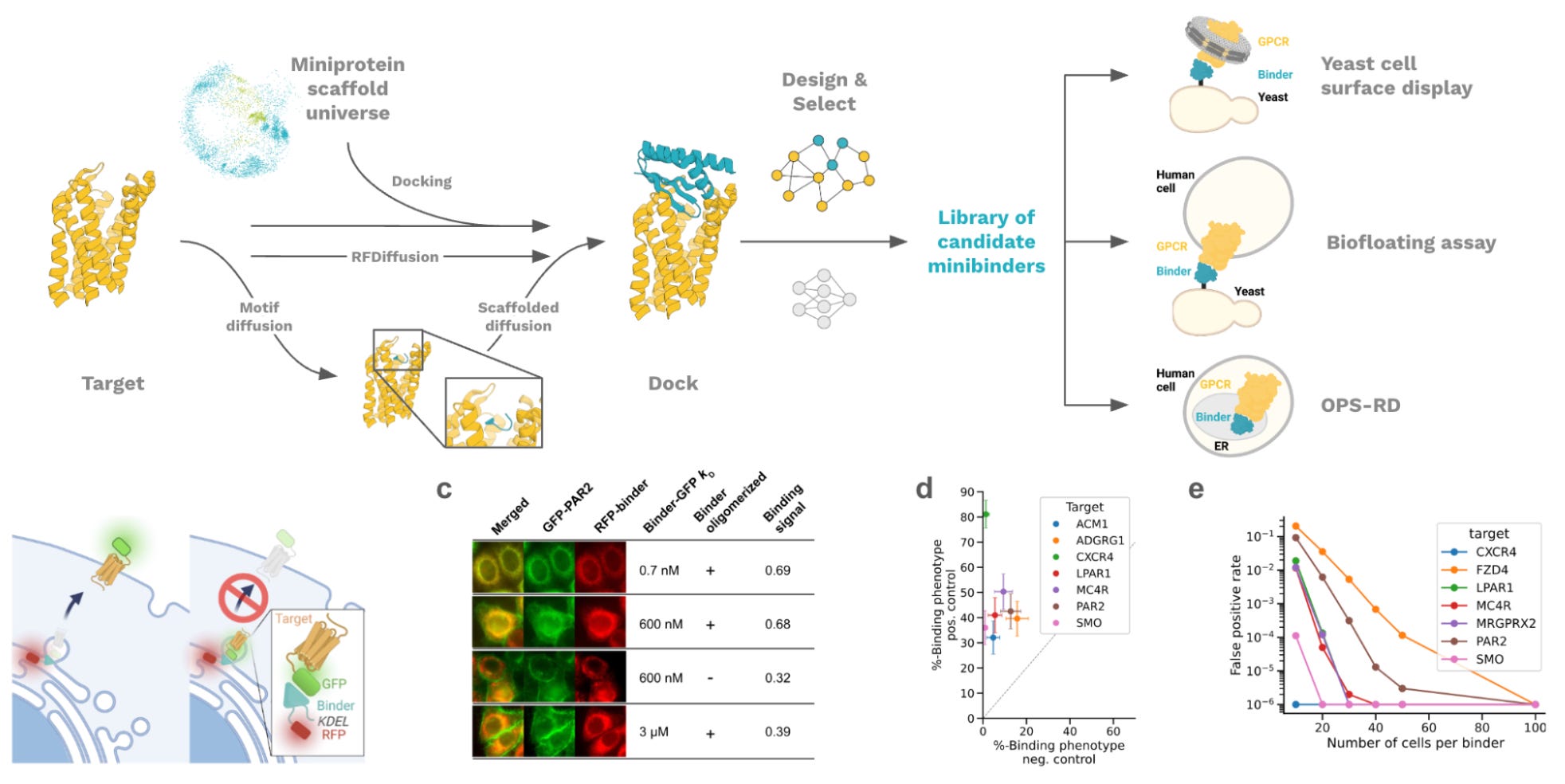
GPCRs are molecular sensors embedded in the cell membrane, where they detect everything from hormones and neurotransmitters to peptides, lipids, and sensory cues. That they are membrane proteins makes them hard to work with experimentally: unlike soluble proteins, they often need detergents, nanodiscs, liposomes, or engineered stabilization to be purified and screened in vitro. They’re also complex functionally - GPCRs undergo a conformational change, which leads to downstream separation and activation of G proteins, which in turn lead to cellular changes. Developing a binder to a GPCR isn’t enough - in order to agonize (activate) the GPCR, you often need to engage specific residues and conformational states buried deep inside the receptor pocket.
Given the deep pockets of GPCRs and the fact that relevant residues for agonism are often deep in the pocket, the team didn’t just throw RFdiffusion at the GPCR to develop binders blindly. To solve this, they first seeded a short five-residue peptide motif into the pocket, letting it make contact with key “hot spot” residues. Once a peptide was discovered in silico that looked good, they used motif-guided RFdiffusion or scaffolds from MetaGen to build a full miniprotein scaffold around it, turning the functional peptide motif into a full minibinder.
But computation alone was not enough. The Skape Bio team also developed a new screening method for asking whether designed binders actually engage GPCRs in a cellular context. Why was this necessary? Because GPCRs are membrane proteins, which makes them awkward targets for many standard display and binding assays. You often need to purify and stabilize the receptor in nanodiscs, liposomes, detergent, or engineered receptor formats, and those workarounds can change receptor conformation or function. Their new approach, Receptor Diversion, turns binding into an optical phenotype inside human cells. The designed binder is retained in the ER using a KDEL tag. As the GFP-tagged GPCR is translated and trafficked through the secretory pathway, strong binders trap more of it in the ER rather than letting it reach the plasma membrane. So instead of only asking “does this purified thing bind this purified thing,” they can image where the receptor goes in the cell, and infer binding from receptor retention.
That readout becomes especially powerful when paired with optical pooled screening. Each design is linked to a DNA barcode, cells are imaged for receptor diversion, and the designs are identified by in situ sequencing. This let them screen libraries approaching 100,000 designs, while still keeping the receptor in a native-like membrane context. They also used more traditional screens where appropriate: yeast display against soluble or nanodisc-stabilized receptor preparations, and a “biofloating” assay where yeast-displayed designs are screened against GPCRs displayed on mammalian cell membranes.
Together, this enabled a broad design-and-screening campaign across 11 GPCR targets. They generated agonists for MRGPRX1 and NK1R, including NK1R designs with nanomolar potency, and antagonists across several class A and class B GPCRs, including CXCR4, CCR5, OXTR, GLP1R, GIPR, GCGR, CGRPR, PTH1R, and PAC1R. The hit rates varied a lot by receptor and assay, but the general pattern is the important part: by combining structure-aware design with high-throughput screening, they were able to design miniproteins that bound and sometimes even activated the previously hard-to-target GPCRs. Still, the work comes with an important caveat. This strategy depends on having useful GPCR structures to design binders to, but designing against “active” or “inactive” states does not automatically guarantee agonism or antagonism - the authors note this as well. Even so, this is a major step toward programmable protein-based modulation of one of the hardest and most important drug target families.
Universal transcriptomic hallmarks of mammalian ageing and mortality [Tyshkovskiy et al., Nature, May 2026]
Why this matters: Aging clocks can accurately predict chronological age, but often provide limited mechanistic insight into what is biologically aging. Most also operate within single species, tissues, or molecular modalities. This paper builds a unified transcriptomic framework spanning mammals, tissues, interventions, and disease states, and argues that aging is not a single process but a conserved, modular architecture of mortality-associated biological programs.
Tyshkovskiy et. al. integrates more than 11,000 transcriptomes across mice, rats, macaques, and humans spanning natural aging, lifespan-extending interventions, progeroid models, chronic disease, and rejuvenation paradigms. Using Gompertz mortality modeling, they construct transcriptomic clocks that predict not only chronological age, but also expected mortality and lifespan modulation, which enables separation of molecular damage burden from chronological time. Across tissues and species, aging consistently associates with upregulation of inflammatory, interferon, p53, and senescence-associated programs, including conserved markers such as CDKN1A, LGALS3, GPNMB, and CST7, while mitochondrial respiration, oxidative phosphorylation, fatty acid metabolism, xenobiotic metabolism, and DNA repair progressively decline. These signatures generalize across bulk tissues, single-cell datasets, and multiple mammalian species, supporting a conserved transcriptomic architecture of mammalian aging.
This paper’s main conceptual contribution is a modular view of aging. Using weighted co-expression network analysis, the authors identify 28 co-regulated modules corresponding to biological subsystems including inflammation, mitochondrial function, chromatin regulation, ECM organization, translation, and mTOR signaling. They then train module-specific clocks, enabling pathway-level quantification of aging dynamics rather than collapsing aging into a single scalar biomarker. This framework reveals that interventions affect aging nonuniformly. Caloric restriction primarily rejuvenates mitochondrial and metabolic modules, while chronic disease preferentially accelerates inflammatory modules. Klotho deficiency strongly accelerates mitochondrial and metabolic aging signatures with surprisingly limited inflammatory activation, suggesting that distinct aging states emerge from different subsystem failures rather than a universal trajectory.
The authors further show that mortality-associated transcriptomic programs are partially reversible. Reprogramming, parabiosis, embryogenesis, and immortalization attenuate or reset multiple aging signatures, while irradiation, senescence, and inflammatory stress recapitulate them. Transcriptomic age acceleration also correlates with DNA methylation aging, particularly in chromatin-associated modules, linking transcriptional and epigenetic aging processes. This work reframes aging as a distributed systems-level process composed of partially separable biological modules. By converting aging hallmarks into quantitative, interpretable transcriptomic clocks, it provides infrastructure for mechanistic aging measurement and pathway-specific rejuvenation studies across species and tissues.
Notable deals
Waypoint Bio raises $20M Series A led by Amplify Partners. Waypoint’s focus is on AI-driven immune drug discovery targeting various solid tumor cancers. To do so, the company leverages pooled screening and emerging spatial biology tools to test the effectiveness of promising molecules. In particular, what differentiates their approach in the ever-more-crowded CAR-T space is their development of novel synthetic protein “armor” for T cells—built by adding and knocking out certain genes. This armor allows the treatment to more effectively target solid tumors, a notoriously more difficult application than blood cancers which have so far experienced greater success. Perhaps most notably, however, is that the funding will go toward carrying out trials for three of the company’s CAR-T candidates in China, where first-in-human trials are much more accessible from both a cost- and time-efficiency standpoint due in large part to their local—instead of national—review boards. Two of the three CAR-Ts entering these trials—targeting pancreatic and gastric cancers—will commence later this year with the third targeting colorectal cancer following at an unspecified future date. Other investors in the round include: General Catalyst, TimeBio Ventures, Lux Capital, and Hummingbird Ventures.
Protuoso Biosciences raises $9.5M seed co-led by Taya Ventures and Darwin Ventures. The company’s hub-and-spoke model integrates well with their core technology, MUXBODIES, described in the press release as “fully recombinant fusion proteins designed to integrate diverse protein modalities, including antibodies, cytokines, peptides, and other signaling modules, into a single therapeutic that can engage multiple biological mechanisms simultaneously.” This technology also helps the company target more complex, interconnected diseases such as obesity, autoimmune, oncology and cardiovascular indications due to the multiple factors associated with their manifestations. Protuoso currently has programs in development for all of the four aforementioned indications, with funding from the round serving to advance these pipelines as well as build out the company’s multifunctional protein engineering platform. Other investors include NSG Ventures, SEEDS, and several further undisclosed participants.
Eli Lilly agrees to purchase vaccine developers Curevo, LimmaTech, and Vaccine Co for $3.8B. Flush with funds from their GLP-1 blockbusters, Lilly has set its sights on a new target: preventing disease at its source. This preventative medicine angle naturally has turned the big pharma’s eye toward vaccines with the simultaneous announcement of agreements to acquire three separate vaccine companies. The first, Curevo, holds a lead vaccine candidate, amezosvatein, targeting shingles. The vaccine includes a next-generation synthetic adjuvant which helps improve tolerability, reducing second dose hesitancy in patients, a major issue facing current shingles prevention. Under this agreement, Curevo and its shareholders could receive up to $1.5B in cash spread across an upfront and unspecified milestone payment. The second company, LimmaTech, has its aim set on bacterial infections experiencing higher rates of antimicrobial resistance, including Staphylococcus aureus, Neisseria gonorrhoeae, and Chlamydia trachomatis. The company’s bacteria toxin and superantigen targeting approach is ideal for generating broad, durable immune responses to complex bacteria, with the pipeline also including other pathogenic targets, particularly those affecting fertility and those with disproportionate effects on women. The deal entails up to $780M in cash, similarly distributed between upfront and milestone payments. Finally, Vaccine Company’s assets consist of In Vivo Nanoparticles (IVN) technologies against viruses, achieving similar results to traditional virus-like particle vaccines (VLPs) but with significant reduction of the manufacturing burden. Though with pipelines spanning multiple viral disease areas, Vaccine Co’s lead is targeting Epstein-Barr Virus (EBV). This final agreement includes payments (both upfront and upon reaching milestones) of up to $1.55B in cash. The underlying thread here is prevention: not just of the primary diseases targeted by the vaccines (e.g. Shingles, EBV) but also of their associated conditions (e.g. stroke, dementia, certain cancers), an admirable goal given the significant positive health outcomes for affected populations potentially yielded by new and innovative preventative health measures.
Perceptic emerges from stealth with $12M in seed funding for end-to-end AI drug discovery.Led by three former executives of Palantir’s Life Sciences team, Perceptic is pitching itself as the “connective tissue” bridging proprietary internal and external data with discrete AI tools, as described in the Fortune coverage. To accomplish this, the infrastructure company is targeting three main areas: scouting biotech-developed assets that show promise for pharma licensing, assisting pharma decision-making regarding selecting assets to progress into clinical trials, and building a “data foundation” for clinical trial design. While many harbor a level of skepticism of AI’s success in biotech with no AI-discovered drugs reaching the market, Perceptic’s CEO, Tillman Flock, cites that the issue lies in the vast majority of AI drug discovery companies targeting only one piece of the development process. Tailored for pharma companies, Perceptic marries and consolidates valuable data from a myriad of sources for its clients, using AI agent workers to selectively hunt and optimize across data domains. The company also maintains a thought-out strategy for avoiding AI hallucinations: their model includes traceability of every claim back to its source. Funds raised from the seed will go toward further engineering buildout and customer acquisition. The round was led by Accel with participation from Air Street Capital and Elder Gull.

In case you missed it
Verge Genomics is rebranding to Verge Labs. Verge Genomics was one of the pioneers applying AI to the target discovery problem in neuroscience. They built CONVERGE, an AI engine trained on human brain tissue, to surface over 280 novel drug targets. This created some partnerships with Eli Lilly and Alexion worth $1.6B and an in-house Phase 1 trial for ALS, which just failed its primary endpoint. Verge has always been a bet that AI can uncover new insights for drug and target discovery, which underwrote the large patient brain tissue transcriptomics data collection and AI model training. They are transitioning to Verge Labs to test out a new business model around this platform: instead of making bets on CONVERGE through their own expensive in-house drug programs, they’re betting on the power of compounding data platform and the increased appetite for big pharma and big AI labs to pay for access to this proprietary data and the AI models enabled by these proprietary data engines.
What we listened to
What we liked on socials channels




Events
Save the Date! Apply here to attend.


Field Trip
Did we miss anything? Would you like to contribute to Decoding Bio by writing a guest post? Drop us a note here or chat with us on Twitter: @decodingbio.