

BioByte 159: the Release of OpenBind, uPAR CAR-T Breaks Down Tumor Defenses, Triplet Tumbling Microscopy Improves Detection of PPIs, and the Potential Role of Autoantibodies in Schizophrenia




Welcome to Decoding Bio’s BioByte: each week our writing collective highlight notable news—from the latest scientific papers to the latest funding rounds—and everything in between. All in one place.


What we read
Blogs
OpenBind’s first release: A structure–affinity dataset for structure-based AI [OpenBind, May 2026]
At the time of its release, AlphaFold3 showed promising capabilities with cofolding (predicting the structure of an entire biomolecular complex at once) including protein-ligand pairs. Naturally, this led to the question of how such tools might bolster structure based drug discovery efforts and replace more computationally expensive workflows for screening compounds and measuring binding affinities. In August 2025, a benchmark called Runs N’ Poses (Škrinjar et al.) demonstrated that AF3 and other cofolding models were essentially memorizing data rather than learning some biophysical mechanism of protein-ligand binding, with performance steeply dropping as input became less similar to the training set. While the development of new architectures might eventually be necessary to create better models, the OpenBind effort was created with the hypothesis that “generating dense, high-quality protein–ligand datasets that link structures with binding measurements” is crucial to improve existing cofolding tools. In this blog post, the OpenBind team outlines their first public dataset release and results showing how it can be used to traditional cofolding methods.
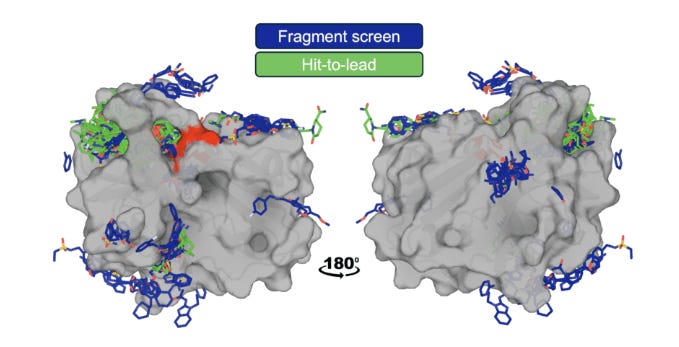
Generally, OpenBind’s data generation efforts are centered around crystallography experiments at the Diamond Light Source synchrotron in the UK. Notably, the group aims not just to increase the overall amount of protein-ligand structural data but to do so in a manner that can most effectively address the current limitations of AI methods. To that end, the group chose Enterovirus A71 (EV-A71) 2A protease as their first target due to its “essential role in the viral life cycle, combined with a druggable protease active site,” making it a promising avenue for antiviral discovery. Over the course of their experiments, the team amassed a dataset of “925 crystallographic binding events from 699 compounds, and associated affinity measurements for 601 compounds.”
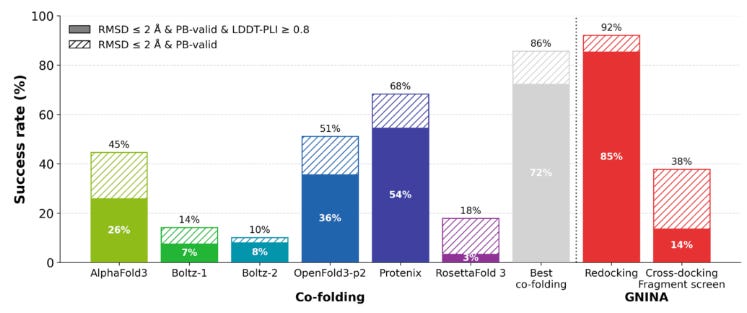
Since EV-A71 2A complexes are fairly dissimilar to structures in the training cutoff for AlphaFold3, the new dataset presented an opportunity to test how cofolding models (Boltz, OpenFold3, and AF3) would compare with traditional docking (AutoDock Vina, GNINA, and DiffDock) and affinity prediction methods on their ability to recover crystallographic poses. The team began by establishing upper and lower bounds of docking tool performance using redocking and crossdocking. In comparatively easier redocking tests (model is given experimentally determined 3D protein structures already conformed to the ligand as input and then has to refit complex), most models performed quite well, reaching success rates above 80%. However, on cross-docking tests (model has to dock ligand into unbound protein structure), performance dropped dramatically (< 5%) due to the challenges of capturing conformational variation in binding interactions. With these baseline, the team found that cofolding models substantially outperformed conventional docking tools on cross-docking due to their ability to model structural flexibility, but still fell short of redocking performance.
Additionally, the team also found that cofolding models could be successfully finetuned using early fragment-screen structures to improve performance. Using OpenFold3-p2 as a platform, fine tuning with EV-A71 2A data boosted success from 36% to 76%. However the team noted that predicting actual binding affinity remained a significant challenge. While Boltz-2 and GNINA showed reasonably high correlations in predictions, most AI-based methods could not surpass basic linear correlation tests between features like a small molecule’s molecular weight and binding affinity (in general, larger molecules are likely to bind better). It will be interesting to see how the continued generation of additional structural data through OpenBind may continue to improve AI-based cofolding models, especially as the consortium explores active learning methods powered by high throughput experimentation.
Treating a new realm of diseases

Beyond traditional limits: explore the drug delivery technology opening new doors in genomic medicine.
This post is sponsored by Cytiva.
Papers
A convergent uPAR-positive tumor ecosystem creates broad vulnerability to CAR T cell therapy [Zhang et al., Cell, March 2026]
Why it matters: Solid tumors have been hard for CAR-T cells to penetrate, with all sorts of efforts to arm CAR-T cells and bolster their persistence as a workaround. Michel Sadelain’s group, which helped pioneer CAR-T therapy for blood cancers, developed a unique targeting strategy to unlock CAR-T efficacy in a broad subset of solid tumors. Instead of only targeting malignant cells directly, they target the broader uPAR+ tumor ecosystem: cancer cells, fibrotic fibroblasts, and immunosuppressive myeloid cells surrounding the tumor. In other words, they take down the “fortress walls” around the tumor while also attacking many of the malignant cells inside. Effective across tumor genetic and tissue heterogeneity, uPAR CAR-T is poised to be a broadly applicable CAR-T therapy across diverse uPAR-high solid tumors.
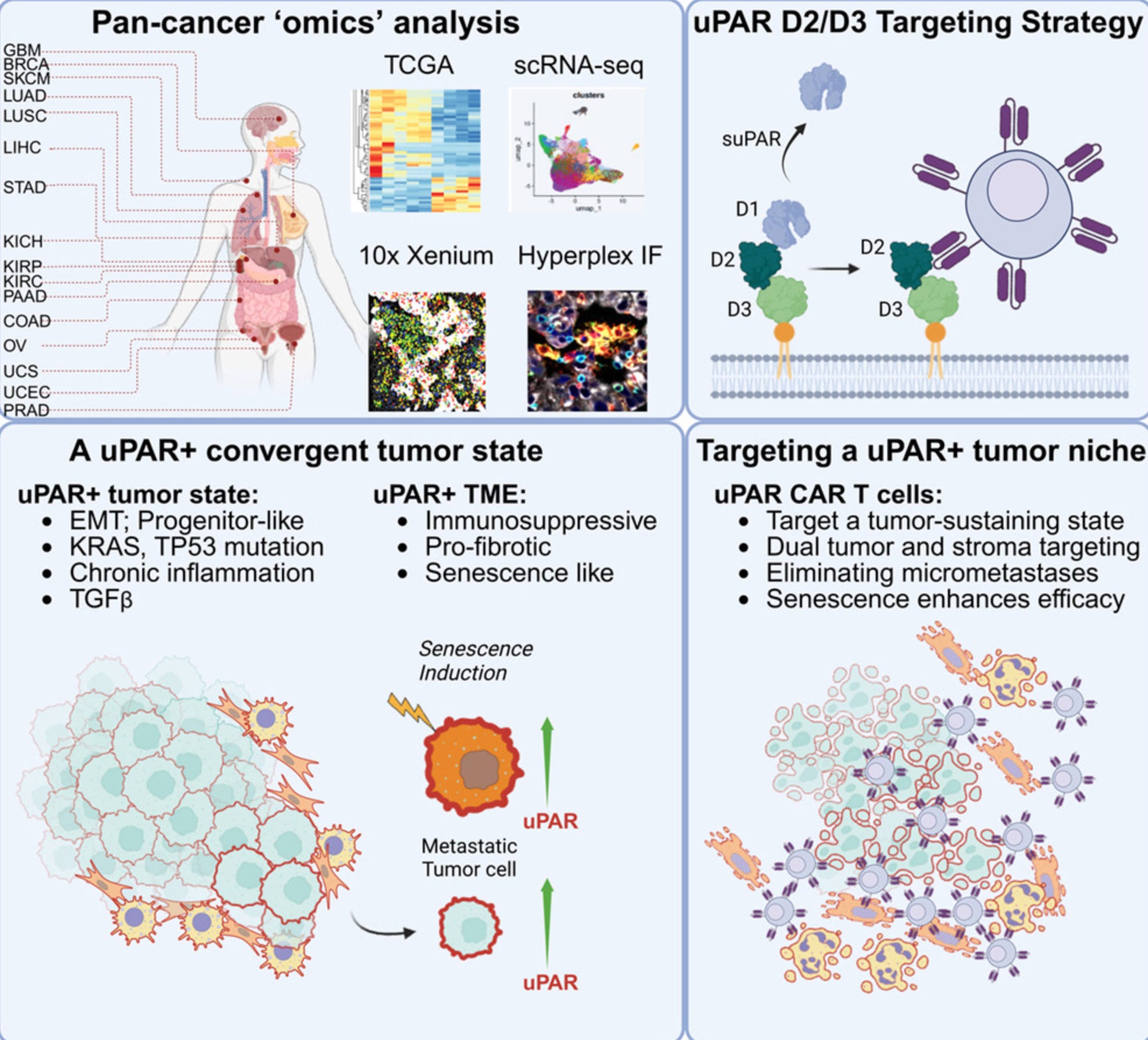
Inspired by their earlier work showing that uPAR CAR-T cells could eliminate senescent cells and reverse fibrosis, researchers from Michel Sadelain’s lab noticed something intriguing: many cancers seem to upregulate this same pathway. Using transcriptomic datasets spanning >10k tumors and >1k patient tumor samples, they found that ~15% of tumors had high uPAR expression. This wasn’t restricted to any single cancer type: it showed up across ovarian, pancreatic, colon, lung, and brain cancers. Importantly, uPAR-high tumors were enriched for TP53 mutations, KRAS/MAPK alterations, EMT programs, fibrosis, and inflammatory signaling, all hallmarks of more aggressive disease. Metastatic tumor cells also had higher PLAUR expression, the gene that encodes uPAR. Alongside these uPAR+ tumor cells, uPAR+ fibroblasts and macrophages formed a fibrotic, immune-suppressive niche around the tumor. This barrier has been one of CAR-T’s biggest problems in solid tumors.
The authors then took their powerful hammer and pointed it at this problem. They developed a uPAR-targeting CAR-T and attempted to clear out uPAR+ tumors. Across multiple lung, pancreatic, and ovarian cancer mouse models, a single infusion drove rapid tumor regression and cleared metastatic disease in ovarian cancer models. During this process, one question arose: were the CAR-T cells killing the cancer cells directly, or were they mainly destroying the surrounding fibrotic scaffold? To answer this, the team built a clever species-mismatched model: tumor cells expressed human uPAR while the surrounding stromal cells expressed mouse uPAR. Using a mouse uPAR CAR-T, which could only directly target the surrounding stromal cells, they showed that stromal targeting alone could reduce tumor burden, while dual tumor-stroma targeting produced more durable control. Breaking down the walls of the castle mattered!
Cellular senescence markers like uPAR also seem to be upregulated after cellular stress. The authors took advantage of this too. Treatments like cisplatin, which damage DNA, pushed tumors into a more senescent-like state, increased uPAR expression, and made the tumors even more vulnerable to uPAR CAR-T. When combining uPAR CAR-T with chemotherapy, 80% of animals remained tumor-free beyond 300 days.
Since uPAR is broadly expressed across cancer subtypes, one natural question that follows from reading the paper is: are there normal, healthy cells expressing uPAR that could be toxically targeted by uPAR CAR-T? uPAR is low or absent in most vital organs, but is detectable on subsets of myeloid cells. To test whether uPAR CAR-T caused myeloid toxicity, the authors looked for changes in blood cell populations after treatment. In an autochthonous ovarian cancer mouse model, uPAR CAR-T cells caused tumor regression with only transient weight loss and no sustained depletion of blood cell populations, suggesting that, in these models, on-target/off-tumor toxicity was limited.
High prevalence of CNS-directed autoantibodies in patients with schizophrenia [Nemani et al., bioRxiv, May 2026]
Why it matters: Anti-NMDA receptor encephalitis showed that autoantibodies (AAbs) (immune proteins targeting the self) can produce a psychiatric phenotype indistinguishable from schizophrenia, but it has been understood as a rare exception rather than a window into mainstream schizophrenia biology. Nemani et al. argue the opposite, that anti-NMDAR encephalitis is reflective of a broader pattern: humoral autoimmunity is a pervasive, structured feature of schizophrenia, where autoantibodies target neural ion channels, synaptic proteins, neuromodulatory GPCRs, and blood-brain barrier components. These reactivities are present near disease onset and accumulate with age. The framing here carves out a candidate immune-driven subset of patients addressable by B-cell depleting therapies the field is already trialing (rituximab is in clinical testing for treatment-resistant schizophrenia regardless of antibody status).
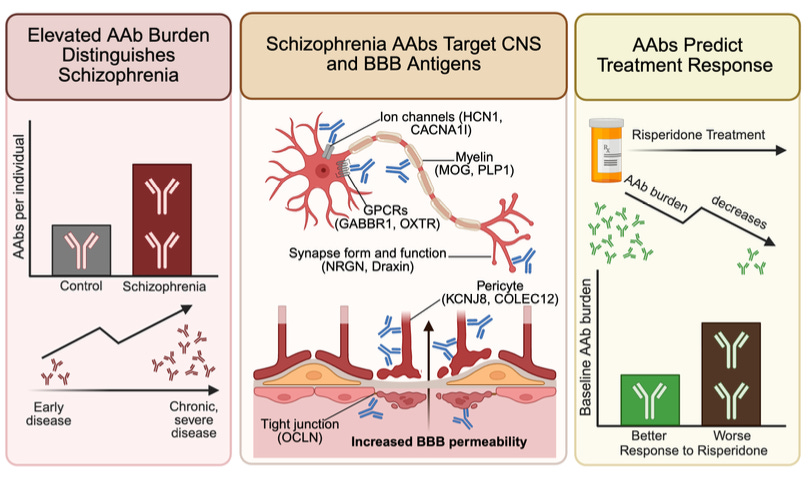
The authors profiled plasma from 352 schizophrenia patients across antipsychotic-naive (at the time of their first psychosis episode), early, and chronic cohorts, with 971 community controls. Using Rapid Extracellular Antigen Profiling (REAP) – a yeast-displayed, proteome-scale screen against secreted and extracellular proteins – they measured AAb reactivity (across the cohort, reactivity to 3,258 proteins was detected). Diagnosis is the dominant correlate of AAb burden in a multivariable regression adjusting for demographics and comorbidities, chronic-psychosis patients carry roughly twice the AAb burden of controls, and a Lasso classifier (regularized logistic regression) separates cases at AUC=0.88. The signal isn’t purely a chronic-illness artifact, given antipsychotic-naive patients show elevated burden at disease onset, and 93% of patient reactivities remain stable across sampling over 3 months.
Of 93 schizophrenia-enriched AAbs, 26 target brain- or neuron-enriched proteins. Encouragingly, hits include HCN1 (a cation channel modulating cortical neurons’ resting potential) and CACNA1I (Cav3.3, a T-type calcium channel controlling thalamic oscillations) – both of which are also schizophrenia GWAS hits.
The blood-brain barrier (BBB) finding is the bridge linking ‘AAbs in the blood’ data to a hypothesis of ‘AAbs are affecting the brain’. 14 of the 93 enriched AAbs target proteins expressed by BBB and brain-CSF barrier-forming cells – brain endothelium, pericytes (contractile cells wrapping brain capillaries), astrocytes (glia that envelope and support the endothelium), vascular smooth muscle, and choroid plexus epithelium (produces CSF and forms B-CSF barrier). In a three-lane microfluidic BBB chip (comprising the first three cell types), plasma from the BBB-AAb-positive [‘BBB(+)’] patients reduces transendothelial electrical resistance (a readout of barrier tightness) and increases permeability to both small and large tracers (biocytin and dextran) relative to BBB-AAb-negative [‘BBB(–)’] plasma. BBB(+) patients also carry ~2x as many brain-targeting AAbs as BBB(–) patients, consistent with a feed-forward model: barrier disruption exposes brain antigens to peripheral immunity and broadens the antibody repertoire over time (potentially creating an autoreactivity flywheel). Clinically, in a 44-patient antipsychotic-naive risperidone trial, higher baseline AAb burden predicted weaker symptom improvement over 16 weeks, and burden itself declined during treatment.
While the cohort signal is robust, the per-antibody molecular story requires further experimentation. The 2 reactivities that were directly tested returned null results: anti-CACNA1I IgG reduced Cav3.3 current density only numerically (not statistically significant) in HEK293 patch clamp recordings, and anti-OXTR IgG did not inhibit oxytocin (a social cognition regulator) receptor signaling in a β-arrestin recruitment assay – a standard downstream GPCR-activation readout. The BBB chip is the strongest functional finding, but uses plasma stratified by AAb status. Purified IgG depletion / reconstitution experiments would be needed to isolate the responsible reactivity and exclude any confounders in plasma. Deeper profiling – including testing CSF in addition to plasma and applying the treatment response prediction to different cohorts – is crucial to contextualizing these findings.
While open questions remain, the team established a rigorous population-level pattern and compelling mechanistic hypotheses, with the signal from REAP converging on known schizophrenia genetics and tracking treatment response. An intriguing question is the extent to which the signature emerges in other psychiatric conditions – whether disease-specific AAb patterns or shared humoral components exist. With further investigation, this work opens up the potential for biological subtyping for a class of diseases that has long resisted it.
Triplet tumbling microscopy enables in situ quantification of protein complex assembly and dynamics [Lazzari-Dean et. al., bioRxiv, May 2026]
Why it matters: Most protein-protein interaction (PPI) measurements either require purified systems or rely on indirect cellular readouts with limited spatial or temporal resolution. Existing live-cell methods face fundamental scaling constraints – translational diffusion changes weakly with complex size, while fluorescence anisotropy is restricted to small proteins because fluorophore lifetimes are too short to capture slower tumbling dynamics. This paper introduces a microscopy framework that extends rotational diffusion measurements into the microsecond regime, which enables direct quantification of protein complex assembly in living cells across a substantially larger size range.
This paper introduces triplet tumbling microscopy (TTM), a live-cell imaging framework that measures protein complex size directly from rotational diffusion (“tumbling”). TTM leverages long-lived triplet states in fluorescent proteins, which persist on microsecond-to-millisecond timescales rather than the nanosecond timescale of fluorescence. The system uses a polarized excitation pulse to generate aligned triplet populations, followed by delayed infrared-triggered emission to measure how much the fluorophores have rotated during the delay period. Because larger complexes tumble more slowly, rotational decay becomes a direct readout of effective complex size.
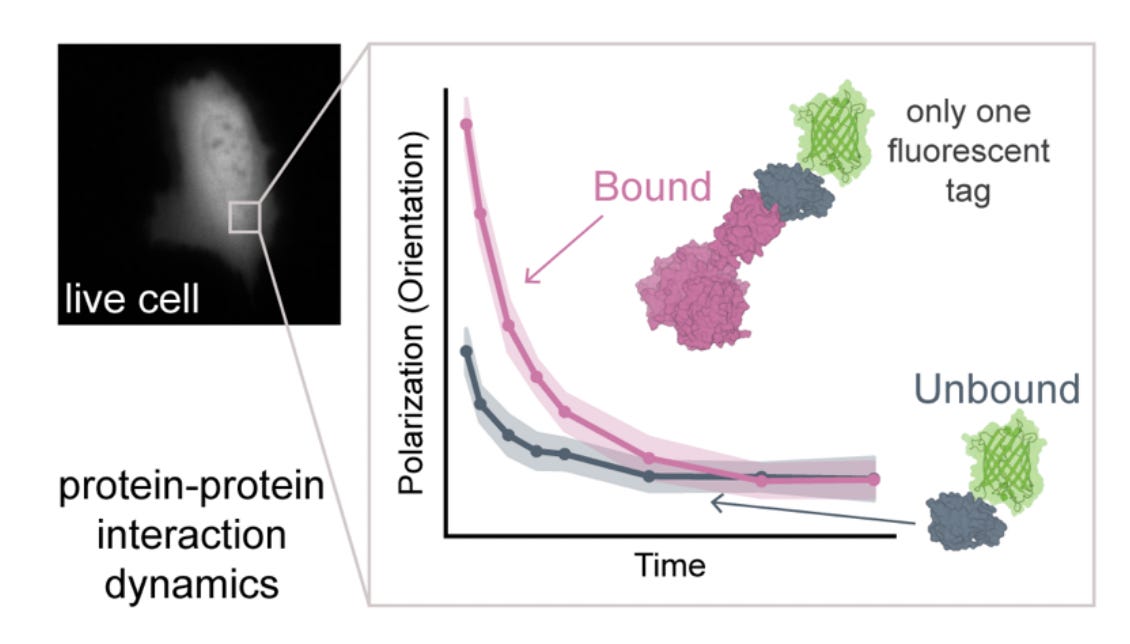
The key advance is extending rotational diffusion measurements into the physical regime relevant for cellular protein assemblies. Conventional fluorescence anisotropy becomes insensitive above ~50 kDa because proteins do not significantly rotate during fluorescence emission. TTM circumvents this limit by using delayed triplet emission as the measurement window, which enables measurements across timescales spanning tens of nanoseconds to hundreds of microseconds. The authors validate this scaling behavior using fluorescent latex beads and show that measured tumbling rates closely follow theoretical predictions from the Stokes-Einstein-Debye relationship.
To establish TTM as a quantitative interaction assay, the authors build a rapamycin-inducible FKBP-FRB dimerization system with binding partners spanning different molecular weights. Rapamycin-induced complex formation predictably slows tumbling, and the extracted decay constants scale linearly with expected complex mass. By mixing bound and unbound populations, they further show that TTM can infer fraction bound through multi-exponential fitting, allowing direct quantification of interaction stoichiometry in heterogeneous samples. The method generalizes to living cells. In U2OS cells, TTM resolves intracellular assembly of complexes ranging from ~41 to ~195 kDa and distinguishes them directly from single-cell cytosolic recordings. Real-time imaging during rapamycin addition captures interaction kinetics on second timescales, showing that TTM can monitor dynamic assembly processes rather than only endpoint states.
The authors then apply TTM to endogenous and clinically relevant interaction systems. For p53, the method resolves differences between monomeric, dimeric, and tetrameric states across a panel of oncogenic mutants, with tumbling measurements correlating strongly with orthogonal crosslinking and size-exclusion assays. Importantly, TTM also detects substantially smaller perturbations. For example, binding of HPV16 E6 to the E3 ligase E6AP increases effective complex mass by only ~10%, yet still produces measurable tumbling shifts in living cells. Mutations that weaken E6-E6AP affinity eliminate the signal, confirming interaction specificity.
TM expands live-cell interaction measurements into a previously inaccessible physical regime. By coupling microscopy with long-timescale rotational diffusion, it enables direct quantification of protein assembly, oligomerization state, and interaction dynamics using only a single fluorescent tag. More broadly, the work reframes rotational diffusion as a scalable in situ readout for macromolecular organization inside living cells.
Notable deals
Isomorphic Labs raises $2.1B Series B round led once again by Thrive Capital. The Google DeepMind spinout builds on original AlphaFold technology with their core product, IsoDDE (Isomorphic Drug Design Engine), which predicts drug interactions and targeting capabilities in the body. Rapidly emerging as a titan in the AI x bio space, Isomorphic already has several ongoing pharma partnerships with leaders Novartis, Eli Lilly and J&J, and made headlines last year with their first external raise of $600M. The previous round was led by Thrive Capital, the same firm leading this colossal new financing, investing alongside other existing participants, Alphabet and GV, with new additions MGX, Temasek, CapitalG, and the UK Sovereign AI Fund also joining the fray. Capital raised will reportedly go toward further honing of IsoDDE and advancing their pipeline as well as expansion of the company’s international operations.
Bristol Myers Squibb announces partnership deal with Hengrui Pharma for the development of 13 early-stage assets worth up to $15B+. Of the 13 assets under the agreement, four are from Hengrui, another four are from BMS, and the remaining five are to be developed jointly utilizing Hengrui’s drug discovery platforms and technology. The therapeutic assets themselves span multiple modalities and indications, namely oncology, hematology, and immunology. Within the partnership, BMS gains global rights to Hengrui’s four assets whilst Hengrui gains rights to commercialize the four from BMS within the “Hengrui Territory” (comprising Mainland China, Hong Kong SAR, and Macau SAR); BMS will however retain rights to their own assets internationally. The fate of the other five assets—seemingly earlier on in the discovery and development process—is more nebulous, with the press release stating potential for a global co-commercialization strategy between the two companies. As part of the deal, BMS will pay $600M upfront to Hengrui plus as additional $125M at the first-year anniversary and again at the second anniversary, though the latter anniversary payment has undisclosed contingencies. Beyond the initial $950M, the remainder of the deal value is comprised of milestone payments, tiered royalties, and options available to be exercised through the joint discovery programs.
Tolemy Bio raises €1.4M in preseed funding led by Norrsken Evolve to build AI-native virtual cell model. The UK-based biotech’s mission centers around a core biopharma bottleneck: “the difficulty of turning complex cell biology experiments into reliable, reusable intelligence.” To overcome this, Tolemy is constructing their core technology, Orbit, which is described as a “control panel for the cell.” Orbit is AI-enabled and fully integrates with scientists’ existing workflows, which is then enhanced by Tolemy’s virtual cell models and paired research agents which help to decode cell responses as well as suggest paths for further experimentation. As such, thorough training and validation of the virtual cell model is a crucial next step and one that relies on the generation of solid experimental datasets—a current focus of the company. Still, the startup has already attracted the attention of MIT and the University of Cambridge, and partnered with GeminiBio to release aiMOS (AI-enabled media optimization service)—which combines AI, ML, and multiomics data for cell therapy manufacturing—in February of this year. In terms of competition amidst the current AI bio arms race, the company is seeking to build and base itself on infrastructure as a means to appeal more to small- and mid-sized biopharma companies—rather than the typical behemoths—who are not capable of the internal infrastructure buildout individually yet still need access to a sophisticated platform for discovery and development. Other investors in the round include Big Sur Ventures, JME Ventures, Masia, and a new UK stealth fund.

What we liked on socials channels



Field Trip
Did we miss anything? Would you like to contribute to Decoding Bio by writing a guest post? Drop us a note here or chat with us on Twitter: @decodingbio.