

BioByte 153: Solving Aging via Induced Proximity Medicines, the Role of lncRNAs in Senescence, Conformational Biasing as a Tool for Protein Engineering, and the Creation of One ESM to Rule Them All




Welcome to Decoding Bio’s BioByte: each week our writing collective highlight notable news—from the latest scientific papers to the latest funding rounds—and everything in between. All in one place.


What we read
Blogs
How to solve aging [Armand B. Cognetta III, Vim, March 2026]
Longevity is one of the hottest new fields and it’s no mystery as to why. With a market that could theoretically include the entire population, a growing number of companies are coming out of the woodwork with a promised focus of “extending the human lifespan.” Yet aging consists of a myriad of underlying diseases and deteriorative processes controlled by complex genomic, transcriptomic, and proteomic relationships and points of failure. As such, a hypothetical cure for aging will need to be sufficiently complex in itself as well as harbor the capability to permeate throughout the body, two shortcomings that are individually faced by various existing modalities. While small molecules are largely able to travel to the far reaches of the human body, they often lack the delicate means to affect complex change; on the other hand, larger molecules such as antibodies, biologics, and gene therapies are equipped to handle such complex interactions but face great difficulty traversing beyond a small subset of the body that characterizes their reachable domain (e.g. the liver, where many large molecules find themselves trapped).
Cognetta thus asserts much of the focus for successful longevity research should be on uncovering new modalities, an area he believes is far too neglected, and that the most logical to pursue is Induced Proximity Medicines (IPMs). Though a newer focus of research, IPMs exist prolifically in nature and have even been unveiled as the underpinnings of several majorly successful drugs such as rapamycin. IPMs work by facilitating the interaction of two macromolecules, generally comprised of a target protein and an effector protein, in a “proximity event” which is how they achieve their desired therapeutic outcome. While they can take the form of either small or large molecules, small molecules are currently favorable given their extended reach as stated above and are thus what Cognetta lasers in on in his post.
As for how they apply to longevity, the answer is through proteostasis and transcription. The ability to maintain proteostasis declines with age which is widespread throughout various bodily systems—from tau tangles and accumulation in the brain associated with cognitive decline and dementia to sarcopenia and immunosenescence—misfolded proteins amass as the body’s natural regenerative capabilities decline. The key here is that proteostasis is also regulated by proximity events, making targeted IPMs an obvious solution. Traveling further upstream, proteostasis, as well as all other aspects of aging, is governed by transcription factors (TFs). Though commonly regarded as undruggable, TFs are still subject to protein-protein interactions. In theory, this implies that through elucidating the compatible partner IPM to control the desired TF—no small feat given the hundreds of thousands of complex interactions between TFs and other proteins—targeted IPM therapeutics could systemically modulate and reprogram aging at its root cause.
This is what Cognetta and team are betting on in their work at General Proximity, a company focused on teasing apart the nuances and potential of IPMs as an anti-aging modality. As the first intentionally developed IPMs, PROTACs (PROteolysis TArgeting Chimeras), are rapidly approaching FDA approval, Cognetta envisions a future in 12 years of an IPM therapeutic, Aevitum, is hitting the markets as the first trillion dollar drug, adding 3-4 years to the average human lifespan and increasing GDP via elevated productivity of working persons taking the drug. Originally developed to treat a rare mitochondrial disorder, its effects in influencing TFs in relation to mitochondrial biogenesis are discovered and harnessed for longevity, making it the first IPM for aging in decades, and the first ever to be rationally designed for this end. While there is still much to unearth within this burgeoning modality, Cognetta insists that the future is bright.
Papers
In vivo generation of anti-BCMA CAR-T cells in relapsed or refractory multiple myeloma: a phase 1 study [An et al., Nature Medicine, March 2026]
Why it matters: This report presents clinical results from one of the first in vivo CAR-T clinical trials, from EsoBiotec. ESO-T01 is a replication-incompetent, immune-shielded lentiviral vector that delivers a humanized anti-BCMA CAR for the treatment of relapsed or refractory multiple myeloma. Four out of five patients achieved objective responses (treatment of the cancer)! However, one patient died from extramedullary lesion-related spinal cord compression, thought to have been worsened by the treatment-driven inflammatory response. Overall, the study highlights both the promise and the unique safety challenges of this new modality.
Multiple myeloma is a blood cancer of plasma cells, the mature B cells that produce antibodies. It has been one of the most successful disease targets for CAR-T therapy. Current approved CAR-T therapies in this space generally target BCMA, a receptor highly expressed in malignant plasma cells. Ex vivo CAR-T therapies such as ide-cel and cilta-cel have shown high remission rates in relapsed or refractory disease (think 70%+ of patients completely defeat their multiple myeloma). However, given the high cost burden (tens of thousands of dollars) and damage induced by lymphodepletion, the search has been on for more scalable CAR-T-like treatments.
Here, the authors tested an in vivo CAR-T approach - in which T-cells within the patient are directly genetically engineered to express the CAR package. ESO-T01 is a replication-incompetent lentiviral vector engineered to generate anti-BCMA CAR-T cells directly in patients, without leukapheresis, ex vivo manufacturing, or lymphodepleting chemotherapy. The vector includes several engineering features intended to improve T-cell targeting and reduce immune clearance, including CD47 overexpression, reduced MHC-I expression, and an anti-TCR nanobody for T-cell-specific targeting. The study enrolled five heavily pretreated patients with relapsed or refractory multiple myeloma, with a median of three prior lines of therapy. The primary endpoint was safety and tolerability, with efficacy evaluated as a secondary endpoint.
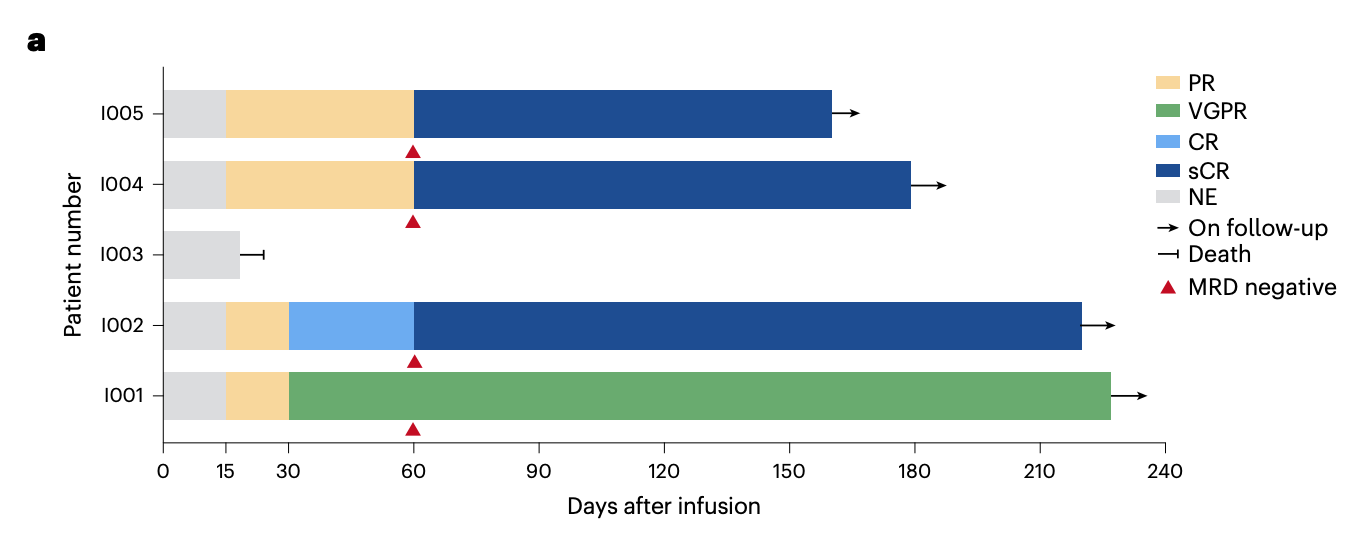
First, the treatment showed clear signs of activity: the overall response rate was 80% (4/5), with responses emerging quickly, at a median of 15 days. By day 60, patient I002 had reached stringent complete response, and patients I004 and I005 had converted from partial response to stringent complete response; patient I001 achieved VGPR and remained there at last follow-up. The fifth patient, who had baseline extramedullary disease involving the spine, died on day 19 from spinal cord compression. The paper notes that this may have reflected inflammatory edema or pseudoprogression associated with CAR-T expansion, although true progression could not be excluded.
Toxicities overlapped with those seen in ex vivo CAR-T therapy, including cytopenias, cytokine release syndrome, and one case of grade 1 ICANS. But the study also points to safety features that may be more specific to in vivo delivery. In particular, the first patient developed a hyperacute inflammatory reaction within hours of infusion, before detectable CAR-T expansion, consistent with an early innate immune response to the vector itself. That event prompted a protocol amendment adding prophylactic dexamethasone before infusion in subsequent patients, which appeared to reduce early toxicity without impairing CAR-T expansion.
Compressing the collective knowledge of ESM into a single protein language model [Dinh et al., Nature Methods, March 2026]
Why it matters: Variant effect prediction models based on protein language models struggle to match the performance of counterparts that incorporate structural and evolutionary information. In this paper, the authors use co-distillation to force an ensemble of PLMs to learn from each other’s high confidence predictions and compress that performance into a single, more efficient variant effect model.
While deep learning-based methods like AlphaFold have made tremendous progress in the field of protein structure prediction, understanding the effect of genetic (and downstream protein) variants as they relate to structure, binding affinity, and larger questions in drug discovery remains a significant challenge. While protein language models (PLMs) have proven quite powerful at learning useful representations of proteins for downstream tasks (including protein structure prediction) for sequence alone, the most performant variant effect prediction (VEP) methods have combined PLMs with more AlphaFold-esque inputs like multiple sequence alignments (MSAs) and protein structures as well as population dynamics data. However, this approach increases complexity, reduces interpretability, and struggles to generalize to proteins that may lack information from the other modalities. In this work, the authors demonstrate that sequence-only PLMs can be significantly improved for VEP with a co-distillation framework and achieve competitive results with more complex approaches.
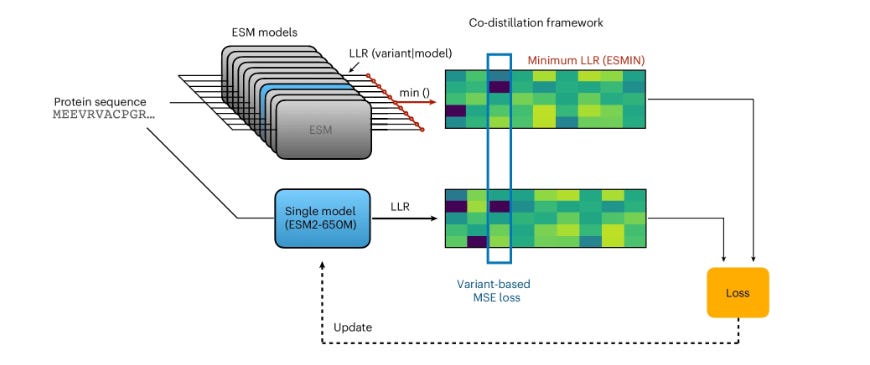
Unlike AlphaFold-style models that are explicitly fed MSAs as input to add evolutionary context, PLMs aim to learn an evolutionary fitness landscape from a series of unaligned sequences. While they still show impressive performance, this approach to training has led to certain deficiencies; for example, the widely used ESM series of models fail to identify conserved structural domains, likely meaning they’d output more false negatives during VEP tasks. Crucially, different ESM models miss different domains despite having relatively similar architectures and training datasets. The authors used this result as the basis for a hypothesis that perhaps the knowledge of multiple ESM models could be condensed into one, and thus developed a multi-step training framework. First, they calculated the log-likelihood ratio for millions of mutations across 11 different ESM models, and recorded the minimum score to establish a maximum confidence metric rather than taking the average which would dilute subtle signals. Next, they used a mutual learning framework, taking that minimum log-likelihood ratio and updating individual ESM models to predict that score using an MSE loss. This was found to significantly increase model performance, after which the authors took the top four most performant models and conducted two more rounds of co-distillation (use inputs from other models as signals to improve your own). Crucially, this second round used average log likelihood since the first round was found to have equalized the variance between pathogenic and benign predictions. This procedure yielded a greatly improved ESM2-3B model that could match the joint performance of the initial eleven model ensemble. Finally, the team distilled the large 3B parameter model into smaller architectures using a teacher-student distillation framework where the smaller models were trained to emulate the output of the large “teacher” model.
Results showed that the new variant ESM (VESM) models powered by co-distillation were able to close the performance gap in a range of VEP tasks. Sequence-only VESM models were able to match MSA- and structure-reliant models like SaProt and PoET, while also beating leading genetic models like AlphaMissense on rare variants. Notably, VESM was able to significantly outperform previous sequence-only models and match MSA-based methods on predicting fitness and activity assays. Some limitations exist - the models are still not very interpretable. Additionally, the memory footprint of some of the model versions can still be quite high. Nonetheless, this paper is an interesting demonstration of how sequence-only models can be improved with new training approaches and help score variants more accurately for downstream analyses.
Multiomic single-cell perturbation screens reveal critical lncRNA regulators of senescence [Zhu et al., Nature Aging, March 2026]
Why it matters: Cellular senescence is a central driver of aging and age-related diseases, yet the regulatory layer controlling it remains poorly mapped. Protein-coding genes explain part of the picture, while long non-coding RNAs (lncRNAs) are widespread but largely uncharacterized. This creates a gap between genomic annotation and fundamental understanding of aging biology.
Zhu et. al. builds an integrative multiomics and CRISPR screening framework to systematically identify lncRNAs that regulate senescence. They combine single-cell RNA-seq, chromatin accessibility, and perturbation data to prioritize candidates linked to aging, DNA damage response, and senescence-associated transcriptional programs.
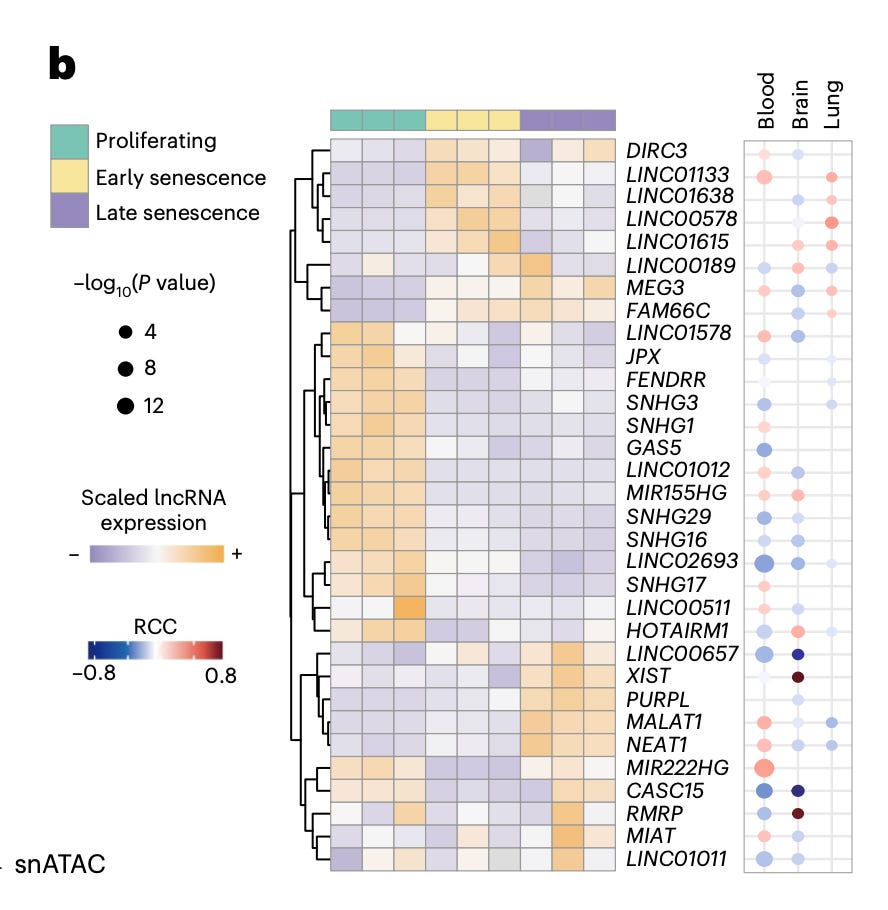
Functionally, they identify a set of pro and anti-senescence lncRNAs. Knockdown of candidates such as MIR155HG, NEAT1, GAS5, and LINC01638 reduces senescence markers and senescent cell fractions, while HOTAIRM1 depletion induces senescence, leading to reduced proliferation and increased expression of canonical markers like p21 and IL-6.
Mechanistically, these lncRNAs act upstream at the level of chromatin and transcriptional regulation, coordinating pathways including DNA repair, inflammatory signaling, and cellular stress responses. HOTAIRM1 in particular links DNA repair and broader cellular programs, placing it as a central node in senescence regulation.
Overall, this work reframes senescence regulation as a distributed non-coding network. Instead of a small set of protein regulators, senescence emerges from coordinated lncRNA-mediated control over chromatin, transcription, and stress pathways, expanding the intervention space for aging and age-related disease.
Computational design of conformation-biasing mutations to alter protein functions [Cavanagh et al., Science, January 2026]
Why it matters: Most natural proteins function by switching between conformational states – a GTPase signals from its GTP-bound but not GDP-bound form, a kinase alternates between autoinhibited and active conformations, a viral spike protein exposes its receptor-binding domain only in the ‘up’ position. Rationally shifting the balance between these states with point mutations would be broadly enabling across areas like molecular signaling or vaccine engineering. However, existing approaches are either too computationally expensive (molecular dynamics), insensitive to single mutations (AlphaFold), or restricted to natural sequence variation (AFCluster). Cavanagh et al. introduce “conformational biasing” (CB), which repurposes inverse folding models (IFMs; predicts amino acid sequences compatible with a given backbone structure) through contrastive scoring: each candidate point mutant is scored against two alternative conformational states, and variants with the most divergent scores are predicted to favor one conformation. CB screens >3,000 variants in under a minute on a consumer GPU, requires no fine-tuning or retraining, and generalizes across structurally diverse proteins.
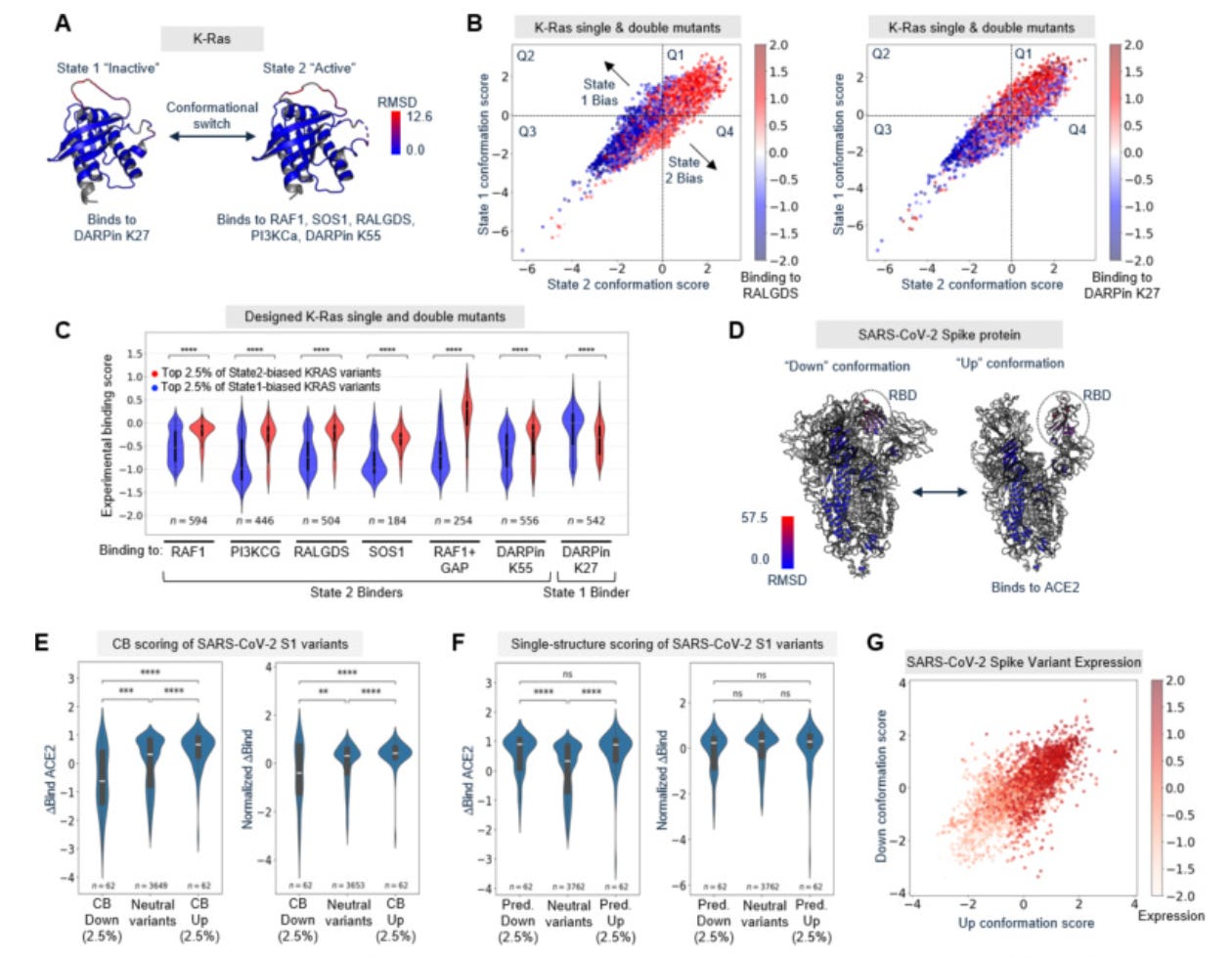
The authors validated CB across seven deep mutational scanning datasets – K-Ras, SARS-CoV-2 spike, the ꞵ2-adrenergic receptor, Src and B-Raf kinases, and two bacterial enzymes (FabZ, MurA). CB-predicted biased variants consistently shifted conformation-specific functions – including effector binding, receptor activation, and enzymatic activity – in the expected directions. A critical control demonstrates the necessity for contrastive scoring: scoring variants against a single backbone structure yielded sets that correlated with expression level (rather than conformational preference) and produced no significant shift in conformation-specific function after expression normalization.
The deepest analysis centers on E. coli lipoic acid ligase (LplA), an enzyme repurposed as a tool for attaching chemical probes (fluorophores, click chemistry handles) to specific target proteins. During catalysis, its C-terminal domain undergoes a 180° rotation – in its ‘closed’ conformation it blocks substrate access to the active site, and in its ‘open’ conformation it exposes the site for probe transfer. The authors directly measured conformational distributions of 16 variants by SEC-SAXS (size-exclusion chromatography + small-angle X-ray scattering; resolves protein shape in solution), achieving a Spearman correlation of 0.81 between CB scores and open-state occupancy – corroborated by an orthogonal tryptophan fluorescence assay across 28 variants. Functionally, 99 variants tested by flow cytometry in HEK293T cells showed that over 90% of CB-predicted open-biased variants had increased promiscuous labeling activity, while over 95% of CB-predicted closed-biased variants showed reduced off-target activity. This uncovered a possible mechanistic model: the C-terminal domain acts as a tethered competitive inhibitor, and shifting the conformational equilibrium tunes the enzyme’s discrimination between intended and unintended substrates. Closed-bias variants reduced background labeling and improved imaging specificity for fluorescent tagging of target proteins in live cells, while open-biased variants labeled hundreds of endogenous proteins in a proximity-dependent manner – laying groundwork for spatial proteomics.
Although CB requires conformational structures in at least two conformational states and has limited recall (identifying high-confidence hits at score distribution tails, not comprehensively across all variants), the speed, accessibility, and breadth of experimental validation position it as a versatile tool for engineering protein dynamics and discovering allosteric mechanisms.
Notable deals
Neion Bio emerges from stealth with $11M seed and first pharma partnership to produce biologics in genetically engineered chicken eggs. Enabled by advances in precision genome engineering and avian primordial germ cell (PGC) cultivation, Neion’s platform, Raptor, re-engineers eggs to produce therapeutic proteins at 10-100x lower cost than conventional Chinese hamster ovary cell manufacturing. Previously considered an uphill battle, egg-based pharma manufacturing has been attempted for 30 years with only one FDA approval (Kanuma, a treatment for a rare liver disorder, in 2016), but the team at Neion argues newer PGC techniques finally make this engineering practical. The biotech’s first brood successfully hatched in September of 2025, with Neion’s numbers now sitting at 50 engineered roosters and presently hatching plans for the next generation of fowl to validate protein expression at useful concentrations. Originally founded in 2024, the company is now stepping into the spotlight with their first commercial deal of co-development and supply of up to three monoclonal antibodies with an unnamed major pharma partner. The seed round was led by Caffeinated Capital with participation from Basis Set and Haystack.
Insilico Medicine signs global R&D collaboration with Eli Lilly worth up to $2.75B. In this deal Lilly obtains exclusive worldwide license for a portfolio of novel oral therapeutics in preclinical development across multiple therapeutic areas, plus collaboration on additional programs using targets selected by Lilly. The partnership pairs Insilico’s Pharma.AI platform with Lilly’s clinical development expertise, adding to the latter’s aggressive AI infrastructure buildout including LillyPod supercomputer and $1B NVIDIA co-innovation lab. In return, Insilico receives $115M upfront, with development, regulatory, and commercial milestones plus tiered royalties. In their press release, Insilico CEO Alex Zhavoronkov emphasizes deploying frontier AI that scales from biomarkers to “life models” to identify multi-purpose targets driving multiple diseases simultaneously, keeping with the company’s devotion to developing deep learning throughout the entirety of the drug discovery process.
Ambrosia Biosciences closes $100M oversubscribed Series B to advance oral small molecule GLP-1 into Phase 1. The company’s cardiometabolic portfolio includes several orally bioavailable candidates targeting key metabolic G-protein coupled receptors (GPCRs), including GLP-1, GIP and amylin. Through a combination of focus on structural biology and advanced computational chemistry techniques, Ambrosia is positioning for a potential best-in-class designation in the highly competitive GLP-1 space. CEO Nick Traggis positions the pipeline within the post-first-generation oral GLP-1 market, emphasizing the company’s small molecules as “designed with combinability in mind.” The round was co-led by Blue Owl Healthcare Opportunities, Redmile, and Deep Track Capital with participation from existing investors, BVF Partners and Boulder Ventures, and new investors, Janus Henderson, Samsara BioCapital, as well as an undisclosed institutional investor.
Centivax closes $37M oversubscribed later-stage financing led by Structure Fund to advance universal flu vaccine Centi-Flu 01 into Phase 2 trials. Centi-Flu 01 targets conserved influenza regions resistant to mutation, aiming to eliminate annual reformulation while protecting against both seasonal and pandemic flu. Phase 2—which will include 500 subjects and is slated to commence in early 2027—is designed to demonstrate superiority against mismatch and currently circulating strains vs. commercial seasonal vaccines. This financing also serves to advance four follow-on programs: a broad cancer therapy, a malaria vaccine, the company’s highly publicized universal snake antivenom (featured earlier this year in Cell), and a broad Alzheimer’s preventative based on CMO Jerald Sadoff’s shingles vaccine work. Other investors in the round include Meiji Seika Pharma (holding first place in Japan’s flu vaccine market share), Sigmas Group, Kendall Capital Partners, and Patrick and John Collison.

In case you missed it
RyboDyn closes $10M seed to decode the "dark proteome" for first-in-class cancer targets.
What we liked on socials channels



Field Trip
Did we miss anything? Would you like to contribute to Decoding Bio by writing a guest post? Drop us a note here or chat with us on Twitter: @decodingbio.