

BioByte 148: MULTI-evolve Tackles Rapid Directed Evolution for Protein Engineering, TerraBind Predicts Binding Affinity, Detectrons Decode Phage Therapy, and Predicting ICI Response in Breast Cancer




Welcome to Decoding Bio’s BioByte: each week our writing collective highlight notable news—from the latest scientific papers to the latest funding rounds—and everything in between. All in one place.

What we read
Papers
Rapid directed evolution guided by protein language models and epistatic interactions [Tran et al., Science, February 2026]
Why it matters: Directed evolution remains the gold standard for protein engineering, but it typically requires many iterative rounds, substantial time, and significant cost. A team led by Vincent Tran at the Arc Institute developed MULTI-evolve, an ML-driven lab-in-the-loop approach for rapidly generating high-fitness multi-mutant protein variants, and demonstrated its performance through benchmarking as well as three engineering campaigns: APEX, dCasRx, and an anti-CD122 antibody.
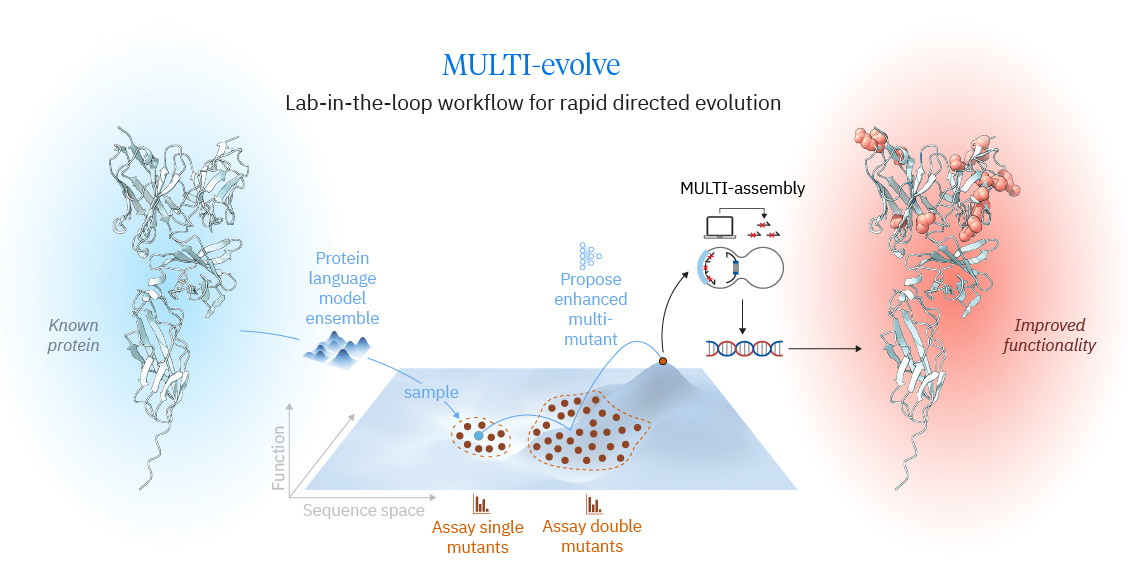
Traditional protein engineering often follows a directed evolution loop: generate a library of variants from the current best sequence, measure fitness, select the best variant, and repeat. The authors aim to compress this cycle using modern machine learning to more efficiently propose high-fitness, multi-mutant designs. Recent approaches have shown that protein language models can efficiently nominate beneficial single mutations, but many of the highest-performing variants require multiple mutations whose effects are not simply additive. As engineers move beyond single substitutions, they must navigate an epistatic landscape with combinations that can help, hurt, or interact in unexpected ways. Tran et al. argue that collecting fitness data for double mutants provides a substantially stronger substrate for modeling epistasis than singles alone, enabling larger jumps across the functional landscape rather than incremental steps.
The MULTI-evolve workflow begins by assembling a high-quality set of candidate single mutations, identified either through experimental screening (for example, deep mutational scanning) or by using ensembles of foundation protein models (for example, ESM and ESM-IF). From these candidates, the authors experimentally measure a compact panel of double mutants to capture interaction effects among mutations, including additive, synergistic, and antagonistic relationships. They then train a supervised model to predict higher-order fitness across mutation combinations and select multi-mutant candidates spanning different mutational loads. To close the lab-in-the-loop cycle, MULTI-evolve is paired with a cloning strategy designed to reliably construct multi-mutant variants without relying exclusively on full gene synthesis.
The authors validate the approach in three distinct engineering campaigns. For APEX/APEX2, designs containing five to seven mutations achieve approximately 193–256× activity gains relative to APEX and still improve 3.6–4.8× relative to the high-performing A134P variant, with multi-mutants outperforming their constituent doubles in a manner consistent with learned synergy. For dCasRx, MULTI-evolve produces five to seven mutation variants with roughly 2.8–9.8× improvements over the wild-type enzyme and increases endogenous trans-splicing by about 3.9–4.5× across three genes. For the anti-CD122 antibody, the authors demonstrate multi-objective optimization in which selected multi-mutants achieve up to roughly 2.0–5.0× combined binding and expression improvements, with follow-up validation showing up to 6.5× yield gains and approximately 2.1–2.7× EC50 improvements.
TerraBind: Fast and Accurate Binding Affinity Prediction through Coarse Structural Representations [Rossi et al., arXiv, February 2026]
Why it matters: State-of-the-art structure predictors are being repurposed for binding affinity, but they’re expensive largely because they generate full 3D all-atom coordinates via a diffusion step. Researchers from Terray Therapeutics argue you don’t need that full coordinate pipeline to predict protein-ligand binding well. They build TerraBind, which runs 26.6× faster than Boltz-2 in their pocket-mode benchmark while outperforming on binding affinity prediction, enabling its use for real-world drug screening campaigns.
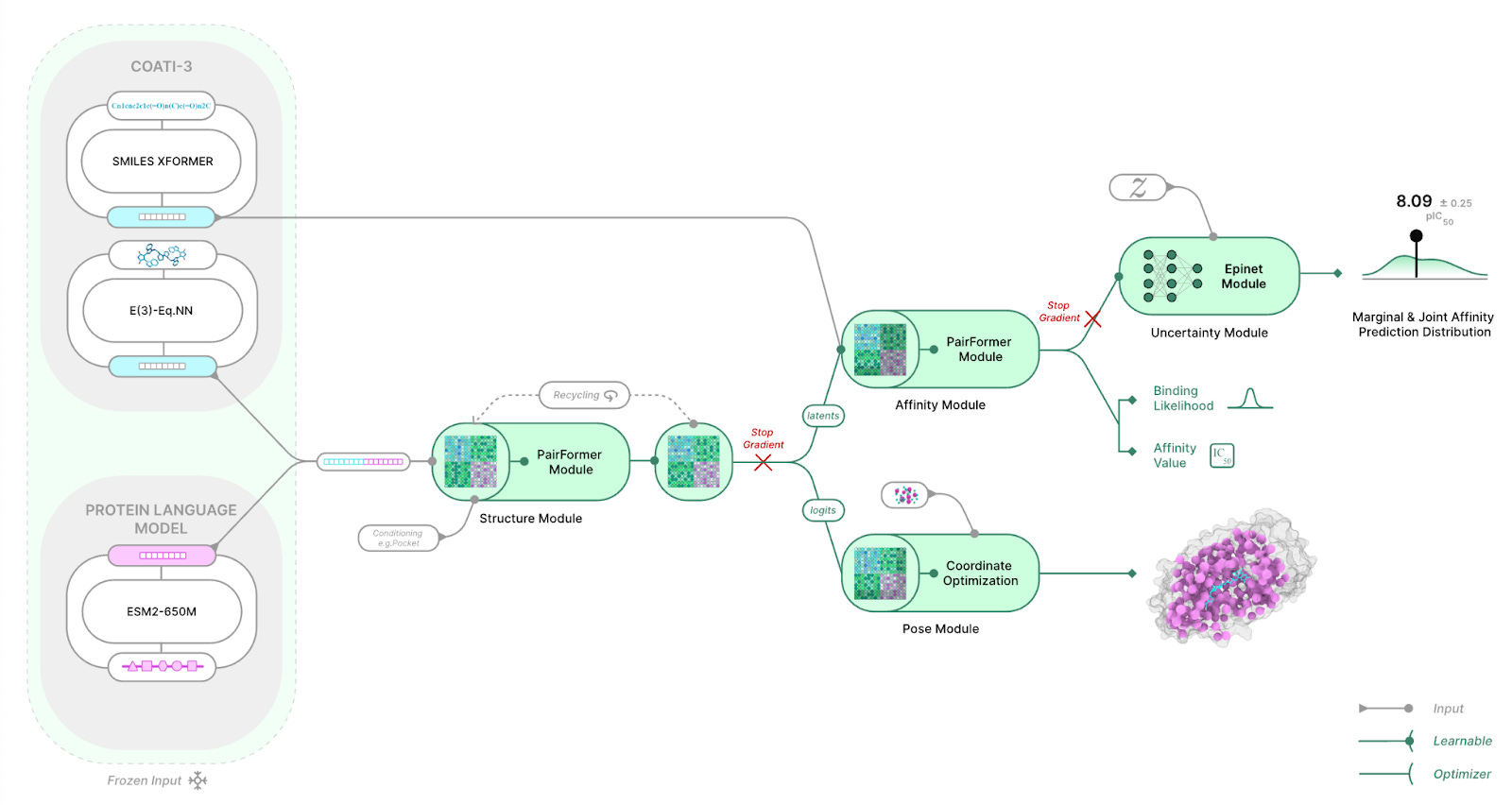
The core bet is that you don’t need full all-atom coordinates to predict binding. To see why, it helps to understand the “distogram”: instead of committing to a single distance (e.g., 5Å), the model predicts a distribution over distance bins for each relevant pair, here ligand heavy-atom ↔ protein residue-center pairs. AlphaFold3-like models then use diffusion to convert these distance distributions into explicit 3D coordinates. The Terray team reasoned that the distogram (and its learned pairwise representations) may already contain most of the signal needed for affinity prediction, so removing diffusion could make the pipeline much lighter and faster without sacrificing accuracy.
Much of the architecture still looks AlphaFold3-like up to the structure trunk. Foundation-model representations of the protein (ESM-2) and ligand (COATI-3) are fed into a Structure module that uses 48 Pairformer blocks to predict the ligand-protein distogram, trained against distograms derived from experimental structures. In Boltz-2, this is followed by a diffusion module to produce 3D coordinates, and those coordinates are then used for affinity prediction. TerraBind skips diffusion and predicts affinity directly from the distogram (and related structural latents) using an additional 6 Pairformer blocks. In total, this reduces trainable parameters from ~509M (Boltz-2) to ~30M (TerraBind), explaining the significant speed gain compared to Boltz-2.
The speedup is large and the affinity gains are where it really differentiates. In their “TerraBind Pocket” setting (196 tokens, 10 samples), they report 1.045s per complex on an A6000 versus 27.8s for Boltz-2, a 26.6× speedup. Structural and pose quality are reported as comparable to Boltz-1, while binding affinity improves: on a proprietary dataset (N=27,078) TerraBind reaches Pearson r = 0.73 versus 0.63 for Boltz-2, and on CASP16 L3000 targets (N=123) TerraBind reports 0.61 versus 0.51 for Boltz-2. They also show that fine-tuning on a small number of new co-crystal structures can further improve affinity performance, and the lighter compute profile makes that kind of iteration much more tractable than Boltz-2, which is exactly what you want when pushing models into large, prospective screening campaigns.
Temporal and spatial composition of the tumor microenvironment predicts response to immune checkpoint inhibition in metastatic TNBC [Greenwald et al., Nature Cancer, February 2026]
Why it matters: Predicting patient responses to immune checkpoint inhibitor therapies for metastatic triple negative breast cancer using traditional biomarkers is still a significant challenge using available biomarker assays. Recognizing that current approaches do not include the greater context of tumor microenvironments, Greenwald et al. demonstrate the use of their SpaceCat pipeline to identify features from longitudinal imaging data to better predict responsiveness.
Immune checkpoint inhibitor (ICI) therapies have shown great promise towards the treatment of skin and lung cancers, but response rates for metastatic breast cancers have been comparatively low. Looking specifically at PDL-1 positive metastatic triple negative breast cancer (mTNBC), chemoimmunotherapy regimes (that combine chemotherapy and immunotherapy approaches) have become the standard of care but understanding which patients will actually respond to the treatment and why remains a key challenge. To that end, current biomarker analyses often fail because they rely on assays that ignore spatial context and the larger complexity of the tumor microenvironment (TME). In response to these limitations, Greenwald et al. detail their analysis of a spatiotemporal TNBC dataset and identify crucial features of the TME landscape at different phases of treatment that may better predict immunotherapy response in patients.
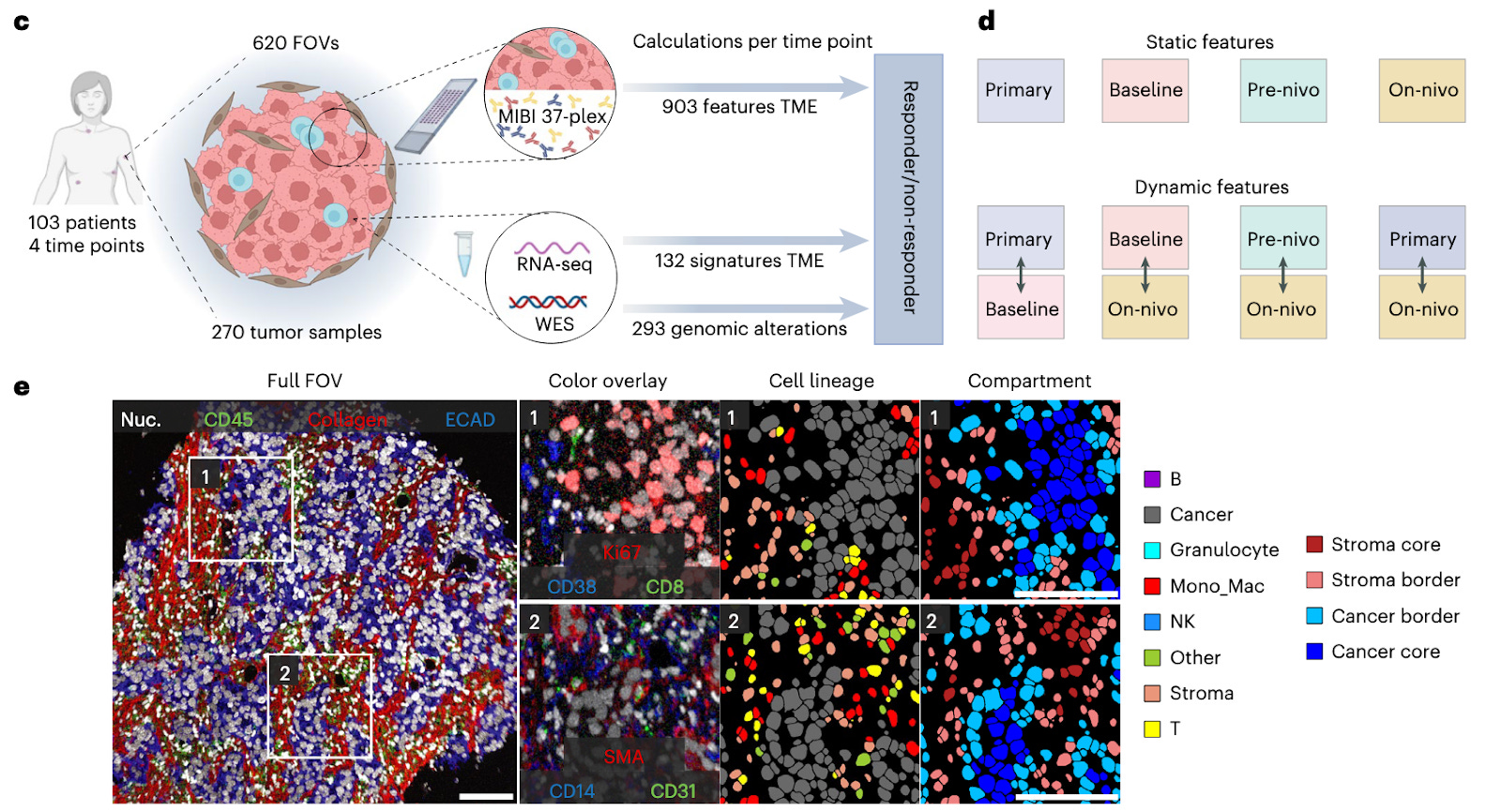
The TONIC trial measures the response of patients with mTBMC and provides researchers with a crucial source of longitudinal data. Specifically, TONIC samples represent four time points in a patient’s disease progression - (1) their primary tumor sometime after identification, (2) a baseline biopsy of a metastatic tumor prior to the start of the trial, (3) prior to treatment with an ICI but after radiation or chemotherapy, and (4) after three cycles of an ICI (in this case, nivolumab). With these four time points, representative regions were chosen by a pathologist to create tissue microarrays (TMAs) for each stage such that antibody panels and multiplexed ion beam imaging (MIBI) could be performed uniformly across patient samples. Using 620 TMA cores, the authors generated multiplexed images for 101 unique patients which were then passed through a cell segmentation (Mesmer to identify individual cells) and classification (Pixie to classify into phenotypes) pipeline to yield labels for 22 cell types representing eight major TME lineages. These images were paired with whole exome sequencing and bulk whole-transcriptome RNA-seq data to complement the information obtained from proteomics readouts and MIBI imaging.
To process this data for further analysis, the authors developed a computational pipeline called SpaceCat that “generates a spatial catalog of informative features from multiplexed image data.” When given an image, SpaceCat generates a set of 903 features like cell density, diversity, spatial interactions, extracellular matrix composition, immune infiltration, etc. First looking into location-specific effects, the authors defined certain tumor compartments - cancer cores, cancer borders, stromal borders, and stromal cores - and then computed SpaceCat features within those compartments and then within a whole image. The vast majority of features were found to be driven by cell phenotype, with a notable dominance of immune-related features (explained by the design focus of the antibody panel). Correlation studies of the features demonstrated the emergence of biologically distinct “modules” related to immune diversity and hypoxia and cell morphology, proving that SpaceCat was capturing coherent signals rather than random noise. Analysis of features at the compartment level and whole-image level showed that over a quarter of features varied significantly across compartments, especially at the tumor-stroma boundaries. Furthermore, cancer core compartments showed significantly higher CD8+/CD4+ T cell ratios pointing to CD8+ cells showing infiltration behavior and CD4+ cells being restricted elsewhere. Before using features to split patients based on response, the authors studied how the TME changed for all patients across the TONIC trial, finding a significant increase in T cell/cancer cell ratios at the stromal border, indicating that nivolumab causes T cell mobilization at the tumor boundary regardless of downstream effect. Other metrics like the ratio of natural killer cells did not show clear separation across time points, suggesting that more nuanced changes might be necessary for clinical benefit.
Moving towards determining the predictive power of SpaceCat, the authors tested every feature to see if it could predict patient response, using an importance score that combined p-value significance and effect size. Their analyses showed a hierarchy of predictors that were most useful for determining survival. Notably, the authors concluded that ratio measurements were more informative than isolated counts features. Additionally, spatial context played a significant role, with cellular diversity at the cancer border being the 31st best predictor at the compartment level but falling to 302nd at the scale of whole images. At a more interpretable level, patients with high ratios of immune cells to cancer cells showed stronger response, which was also strongly linked with PD-L1 expression on macrophages and antigen presenting cells. High cellular diversity was consistently predictive of success while monoculture tumors tended to fail. Interestingly, features like cancer-immune mixing (measure of immune cell penetrance into the tumor) seemed to have little predictive value for mTBMC despite being a key predictor for primary TBMC. Additionally, proliferation markers and hypoxia did not add much value as well, reinforcing the idea that metastatic TBMC is markedly different from primary TBMC and that biomarkers cannot be recycled between the two diseases. Interestingly, the most predictive features of the study were only predictive at a specific time point, with the majority of power coming from features derived at the treatment stage. This suggests that the state of the tumor prior to treatment is far less informative than information from the tumor shortly after the initial doses. Overall, this study reaffirms the idea that metastatic breast cancer is significantly different from its primary counterpart, and that spatial approaches like SpaceCat can help overcome the limitations of traditional biomarkers when predicting patient response to treatment.
Systemic hypoxia suppresses solid tumor growth [Midha et al., bioRxiv, February 2026]
Why it matters: Tumor hypoxia is typically treated as a hallmark of aggressive disease and therapy resistance, but most evidence concerns local hypoxia within poorly perfused tumors. This paper isolates the systemic variable and shows the opposite effect: whole-body hypoxia suppresses solid tumor growth. The implication is that oxygen is not just a constraint on tumor physiology but also a host-level input that can be therapeutically modulated, potentially through drug-like approaches.
Midha et al. show that lowering inhaled oxygen (11% or 8% O₂) significantly reduces tumor growth across multiple models, including syngeneic Panc02 pancreatic tumors, orthotopic E0771 breast tumors, and a genetically engineered KPC pancreatic cancer model. Direct measurements confirm that systemic hypoxia reduces intratumoral pO₂ while still producing slower tumor expansion.
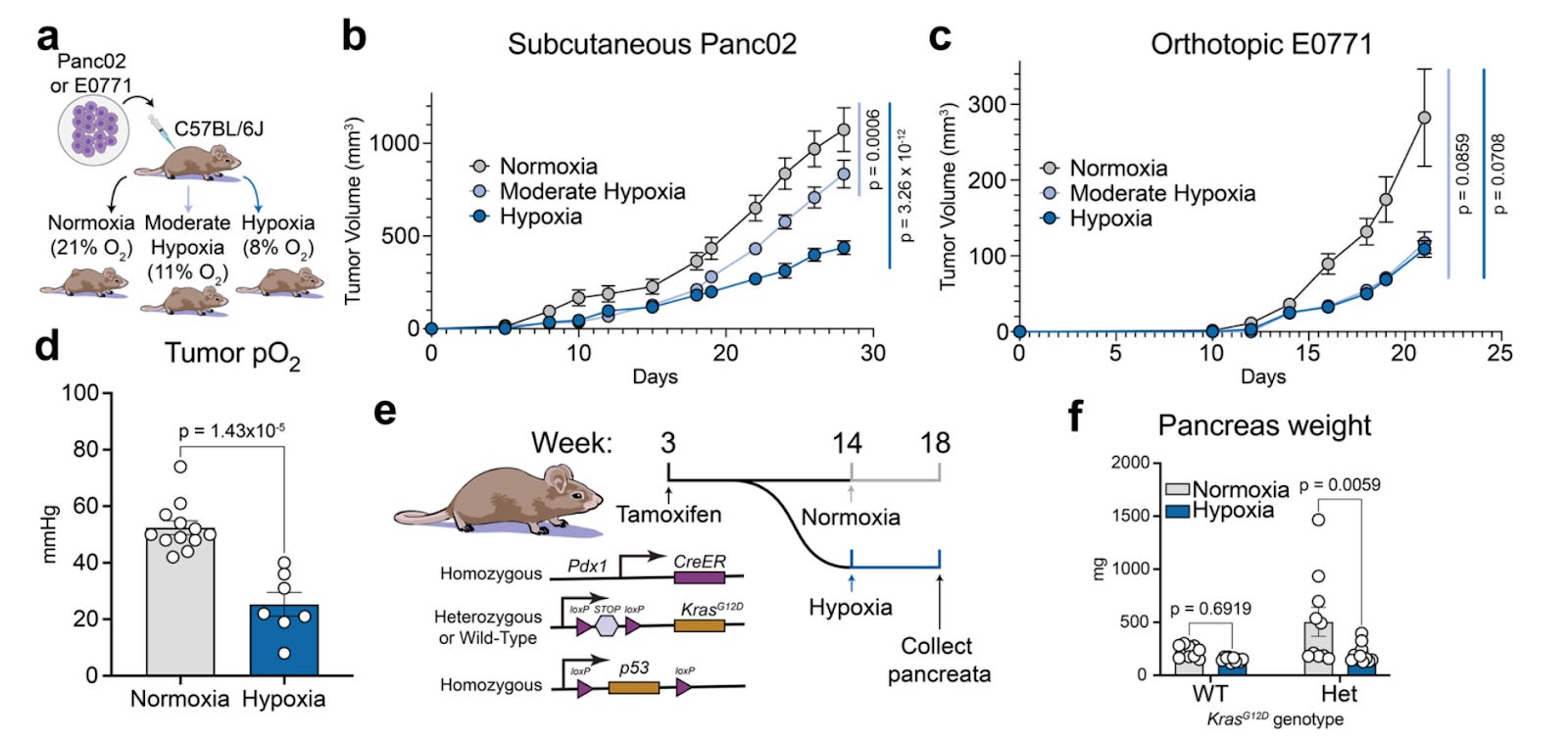
To test generality across lineages, the authors use GENEVA, a pooled xenograft system of 20 human cancer cell lines in the same host. Systemic hypoxia reduces overall tumor burden but reveals heterogeneous sensitivity. Most lineages show reduced fitness and G1 arrest, while a small subset (including renal carcinoma lines) exhibits relative resistance. Several systemic explanations are ruled out. Hypoxia induces hypoglycemia, but tumors increase FDG uptake and GLUT1 expression, and glucose supplementation does not rescue growth. Constitutive insulin signaling via PTEN loss does not eliminate hypoxia sensitivity. Canonical HIF signaling is also not required: ARNT (HIF-1β) knockout tumors remain suppressed, and HIF target induction does not correlate with fitness.
Instead, metabolomics and tracing converge on a nucleotide bottleneck. Hypoxic tumors show broad depletion of nucleotide pools and altered purine intermediates. Dual isotope tracing (¹⁵N-glutamine and ¹³C-adenine) demonstrates that systemic hypoxia suppresses de novo purine synthesis and increases reliance on salvage pathways, both in vitro and in vivo. Consistently, hypoxia-sensitive lineages downregulate key purine synthesis genes (PPAT, MTHFD1, PAICS, ATIC), while salvage pathway expression does not track with response. Therapeutically, systemic hypoxia shows sustained efficacy and compatibility with standard treatments. Tumors do not develop resistance across serial passaging, and hypoxia synergizes with gemcitabine and anti-CTLA4 immunotherapy. The authors also demonstrate a pharmacological implementation using HypoxyStat, a small molecule that increases hemoglobin oxygen affinity to induce systemic hypoxia and suppress tumor growth.
Overall, this work reframes hypoxia as context-dependent. Local hypoxia may promote malignancy through spatial division of labor, but systemic hypoxia collapses anabolic capacity by constraining nucleotide synthesis. The core implication is that host oxygenation is a tunable metabolic lever, with purine synthesis as a central vulnerability for suppressing solid tumor growth.
Programmable RNA detection generates DNA barcodes for multiplexed phage-host interaction screening [Han and Shipman, bioRxiv, February 2026]
Why it matters: Phage therapy is resurging as antibiotic resistance climbs, but identifying which phage infects which bacterial strain remains a slow, low-throughput process. Most RNA sensors report via fluorescence or growth, making deep multiplexing in pooled cultures difficult. Han and Shipman introduce ‘Detectrons’ – programmable RNA switches that convert the presence of a specific RNA into a sequence-encoded DNA barcode – allowing phage infections and host susceptibility to be read out by sequencing in a single mixed culture. This shift – from transient RNA sensing to durable DNA memory – lays the groundwork of pooled, massively multiplexed assays where cellular responses are digitally recorded.
Detectrons integrate a toehold switch with a retron (bacterial genetic element producing ssDNA from its RNA template) noncoding RNA so that detection of a target RNA transcript triggers reverse transcription (RT) of a unique DNA barcode. In the ‘OFF’ state, the toehold sequesters the retron’s a1/a2 complementarity region, blocking RT; cognate RNA restores this structure via strand displacement, initiating barcode synthesis.
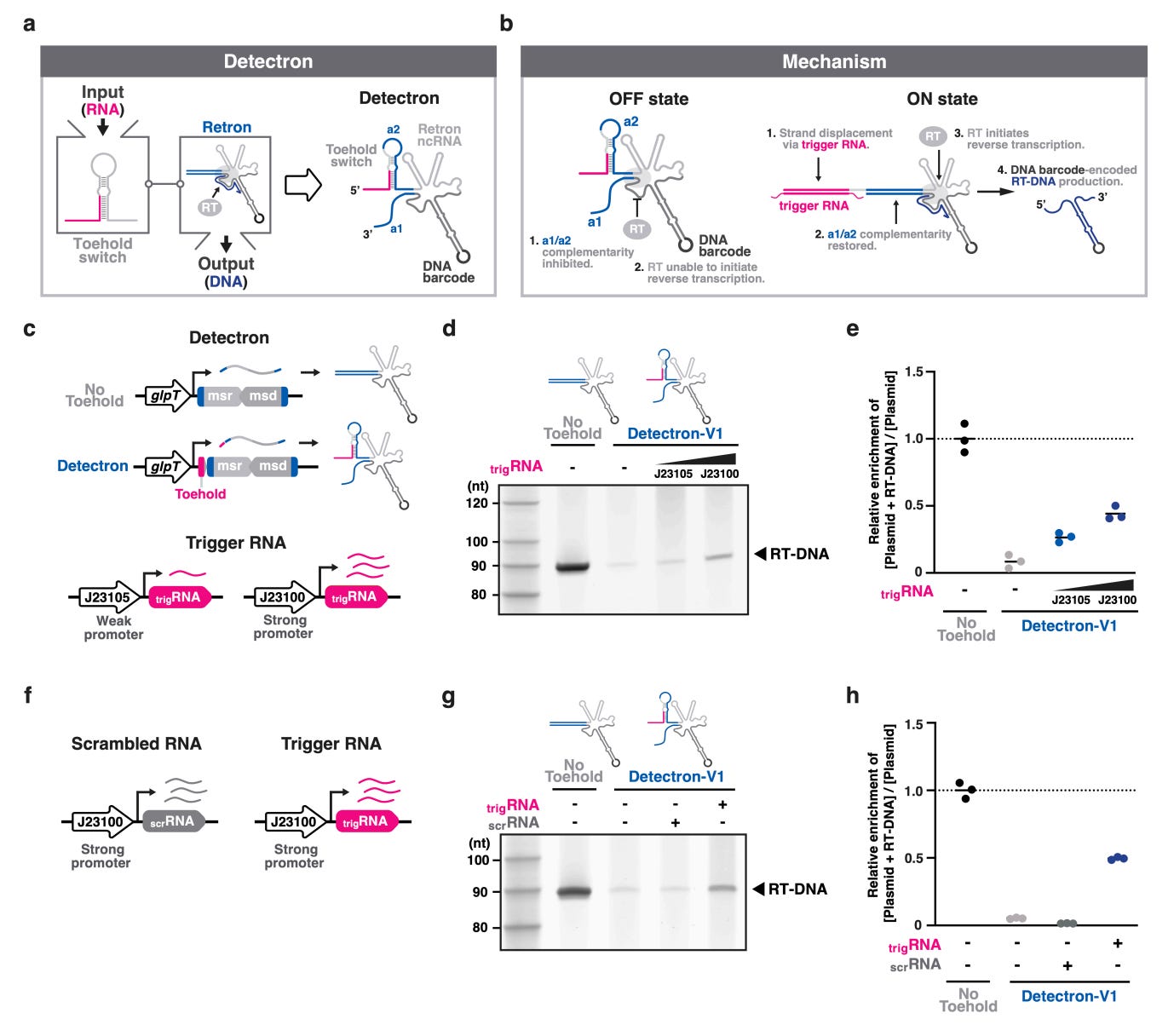
The initial Detectron prototype was created by grafting a pre-existing Zika-targeting toehold switch onto retron-Eco1 – a functional approach, but one with limited dynamic range. To generalize and optimize the design, the authors constructed a library of 7,840 toehold-retron variants (yielding 23,520 barcoded constructs) – systematically shifting structural parameters of the toehold switch. They screened for ‘ON’ / ‘OFF’ ratios (fold-change in barcode production with / without trigger RNA) by sequencing, and trained an XGBoost Model (r2 = 0.794) that identified key structural determinants of the toehold-retron hairpin that govern switching efficiency – combined stem length of 10-11 bp, no bulge, 12 bp loop, and 1bp distance to the reverse-transcription priming guanosine. The top variant (#621) achieved a 14.7-fold improvement in ‘ON’ / ‘OFF’ ratio over the original design and retained its performance in K. pneumoniae and C. freundii, demonstrating cross-species portability.
Applying the optimized Detectron-V2 architecture to phage-host analyses, the authors built sensors targeting transcripts from phages T4, T5, and T7, each linked to a distinct barcode. In pooled E. coli cultures challenged with a single phage, only the cognate barcode was induced, enabling unambiguous identification of the infecting phage from a single sequencing readout. A dual-barcoding extension – encoding both phage identity and host strain identity – allowed quantitative profiling of T7 infection susceptibility across wild-type and LPS-mutant E. coli strains in one assay, with barcode signal intensity tracking the expected gradient of receptor-mediated adsorption efficiency. Detectrons are the first system to directly transduce RNA inputs into DNA outputs, and their modular, sequence-programmable design positions them as a scalable platform for phage screening and, more broadly, for recording transcriptional events in complex microbial communities.
Notable deals
Korsana Biosciences emerges from stealth this week, carrying with them $175M in capital raised since their founding in 2024. The funds include a 2024 seed round of $25M from Fairmount and Venrock Healthcare Capital Partners and a $150M Series A from September of 2025 led by TCGX and Wellington Management. Other investors in the Series A included Foresite Capital, Janus Henderson Investors, J.P. Morgan Life Sciences Private Capital, and Sanofi Ventures. With an expected runway into 2028, Korsana will use the two rounds to fund their lead program, KRSA-028, through the early stages of clinical trials, as well as furthering the development of their earlier programs. The lead asset is a shuttled antibody from a partnership with Paragon Therapeutics that is designed to treat Alzheimer’s disease by targeting amyloid beta, built using the company’s proprietary Therapeutic Targeting (THETA™) platform which helps improve brain delivery. Data demonstrating plaque clearance in Alzheimer’s patients is expected in late 2027.
Sensei Biotherapeutics announces both the successful acquisition of Faeth Therapeutics in a stock-for-stock transaction and a $200M Series B through private placement financing. The two announcements are complementary, as the acquisition brings Faeth’s lead oncology asset, PIKTOR, into Sensei’s pipeline while the additional capital provides sufficient funding for the ongoing Phase II trial of the asset in advanced endometrial cancer as well as starting a Phase Ib trial for breast cancer. PIKTOR is a combination therapeutic of sapanisertib and serabelisib, designed to robustly inhibit the PI3K/AKT/mTOR pathway. Investors in the private placement financing included B Group Capital, Balyasny Asset Management, Columbia Threadneedle Investments, Cormorant Asset Management, Fairmount, Logos Capital, RA Capital Management, and Vivo Capital.
Gilead acquires the global rights to Genhouse Bio’s synthetic lethal therapy, GH31, for $80M upfront with an additional $1.45B in milestones. The China biotech has already been approved to take the asset, which inhibits MAT2A, into clinical trials for various cancers in both China and the US with IND approval. Late last year, Gilead acquired another synthetic lethal therapy from Repare Therapeutics for $25M.

What we listened to
What we liked on socials channels



Field Trip
Did we miss anything? Would you like to contribute to Decoding Bio by writing a guest post? Drop us a note here or chat with us on Twitter: @decodingbio.
Great summary. What I like about MULTI evolve is that it quietly reframes directed evolution as an experimental design problem. Instead of more rounds, you buy information efficiently: pick singles, sample a minimal set of doubles that maximizes epistasis signal, then let the model propose the multi mutant. It feels like the protein engineering version of learning the interaction terms in a sparse model, which is exactly what rugged landscapes demand.