

BioByte 144: A Virtual Model of Cell Signaling, Mapping Aptamer-Protein Binding with SPARK-seq, Immunogenicity Prediction with ImmunoStruct, and Sleep as a Systemic Biomarker




Welcome to Decoding Bio’s BioByte: each week our writing collective highlight notable news—from the latest scientific papers to the latest funding rounds—and everything in between. All in one place.


What we read
Blogs
Engineering Cell Fate: Towards a Foundation Model for Virtual Cell Signaling [Cellular Intelligence, January 2026]
Although there are only about 20 known fundamental molecular signaling pathways that cells utilize to communicate with each other, when employed together, they give rise to an immense diversity of cellular states. This complexity is due to the dependence of the cell’s final state on the initial state as well as the combination, order, and dose of signaling molecules. The variation resulting from initial cell state (context dependence) taken with the quantity of possible perturbations (combinatorial complexity) has historically resulted in an inability to predict cellular behavior, resulting in time-consuming, trial-and-error experimentation when trying to determine the effect of a specific signal.
Building a concrete understanding as to how these signaling pathways dictate cell state in healthy cells—and how they are disrupted or dysregulated in diseased cells—is imperative to the acceleration of regenerative medicine and pharmacology. In pursuit of this aim, Cellular Intelligence outlines their current approach to building a Universal Virtual Cell-Signaling Model, one rooted in vast amounts of experimental data with the aim of being able to accurately predict the outcome state of a cell based on the initial state and perturbation.
“In short, the model answers the question: ‘Given this type of cell (stem cell, cancer cell, neuron) and this signal at this dose, what will the cell look like and do next?’”
Cellular Intelligence takes a unique approach to building their underlying datasets. The primary limitation they have identified is a lack of data that is both context-rich and perturbation-rich. As such, their experimental processes ensure both sufficient variety of the signal and initial cell state by leveraging the inherent ability of pluripotent stem cells to differentiate, combined with their ‘split-and-pool’ approach. Using barcoded capsules, they are also able to track each cell’s unique treatment.
The split-and-pool approach allows for an exponentially expanding set of conditions, facilitating rapid, diverse data generation. Once each capsule is treated with a stimuli, it is split according to the number of unique perturbations to be applied in the subsequent step. For N stimuli applied in k rounds, this results in a scaling of Nk, so 30 stimuli across three rounds would produce 27000 unique sequences. Cellular Intelligence has already successfully been able to scale this process to 1M unique conditions.
The data generated from their experimental pipeline, and eventually public datasets, is fed into their transformer model, which akin to how ChatGPT seeks to predict the next word in a sequence, seeks to predict the next cellular state after a perturbation. The model is also able to help inform subsequent experiments by identifying gaps in its understanding and proposing the most valuable combinations to test.
Although they are still in their self-defined Alpha Phase, in just two years the company has gone from concept to validating their approach and executing a 1M-condition run. The insights afforded by their model will have a number of impactful applications, including facilitating rational design of cell differentiation protocols in addition to enabling understanding signaling failure in disease, treatment of signaling differences arising from genetic variation, and prediction of diseased cell response after drug exposure. These benefits have the potential to vastly accelerate answers to questions such as “How do I rescue this diseased cell?”, bringing treatments to patients without the need for excessive years of manual optimization.
Papers
SPARK-seq: A high-throughput platform for aptamer discovery and kinetic profiling [Luo et al., Science, January 2026]
Why it matters: Luo et al. cleverly design a CRISPR-based, high throughput experiment to map DNA aptamers to the proteins that they bind to - enabling generative design of target specific aptamers.
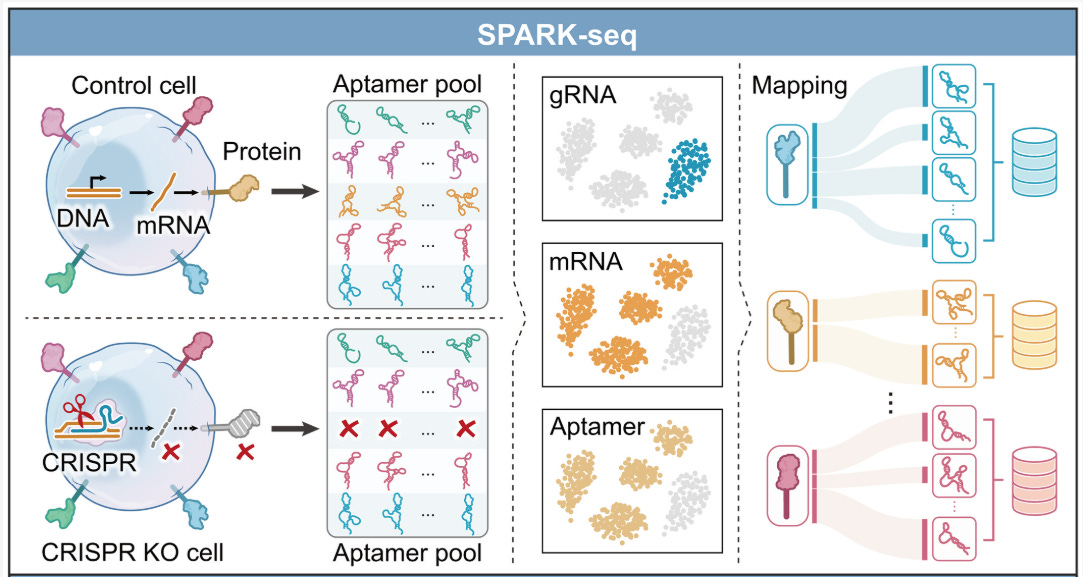
Aptamers are short single-stranded DNA or RNA molecules that fold into structures capable of binding molecular targets with high specificity. Using SELEX, scientists can iteratively enrich large random libraries to obtain binders to specific molecular targets, which are then actively deployed as probes, diagnostics, imaging agents, and targeting ligands. However, standard SELEX often yields binders without knowing the exact protein target or binding mode, meaning we are waiting for a good DNA aptamer to fall through a sieve and into our laps. As seen in the protein design field, large-scale high-fidelity molecular data like the PDB combined with generative machine learning enabled scientists to not just rely on selection but actually design binders towards specific targets. However, DNA aptamers don’t have this enabling dataset mapping DNA aptamer sequence with target protein identity.
SPARK-seq addresses this by combining Cell-SELEX with pooled CRISPR knockouts and single-cell sequencing to generate a dataset of interactions between DNA aptamers and cell surface proteins. After enriching an aptamer library on live cells for tight binding aptamers, the authors expose the library to a mixed population of cells in which individual surface proteins have been knocked out using CRISPR. They then use single-cell sequencing to identify, for each cell, what cell surface protein has been knocked out, what DNA aptamers are bound to the cell, and the transcriptome. The intuition here is simple: if knocking out protein X causes a specific aptamer family to stop binding a cell, that family likely binds protein X. Because the binding signal reflects how long aptamers remain associated with cells, the resulting data is strongly coupled to dissociation kinetics (koff), which is awesome for diagnostic and therapeutic aptamer applications.
With these target-labeled aptamer families, the authors train sequence-based ML models separately for each protein: a convolutional neural network that learns to classify whether a nucleotide sequence binds a given target, and a generative model trained on known binders to propose new sequence variants. New aptamers are generated from the target-specific sequence distribution and then evaluated by the classifier, enabling in silico optimization before experimental validation. They validate predicted interactions using flow cytometry and biophysical assays, and show that SPARK-seq recovers binders to low-abundance and conformationally sensitive targets that are often missed by pull-down or mass spectrometry–based methods.
ImmunoStruct enables multimodal deep learning for immunogenicity prediction [Givechian et al., Nature Machine Intelligence, December 2025]
Why it matters: Givechian et al. aim to overcome a key bottleneck in the development of immunotherapy vaccines for infectious disease and cancer by predicting the immunogenicity of peptides. ImmunoStruct leverages sequence, structural, and biochemical information to achieve substantial performance gains over sequence-only models and is a hopeful step towards improved vaccine design capabilities.
Epitope-based vaccines are a promising frontier in immunotherapy for the targeted treatment of viruses and cancers. However, current delivery methods like lipid nanoparticle platforms can only carry a few peptides, crucially limiting the number of candidates that may elicit an immune response. Additionally, patients themselves show low response rates, with some studies showing that only 2-6% of mutation-derived peptides induced genuine T cell activation and immunogenicity. This challenge is also compounded by the complexity of the human immune system. Therefore, one key challenge in this space is improving the identification of immunogenic peptides that can improve the overall effectiveness of epitope-based vaccines. In this paper, the authors present ImmunoStruct, a multimodal deep learning model with accompanying wet lab validations studies that predicts class I peptide-MHC immunogenicity.
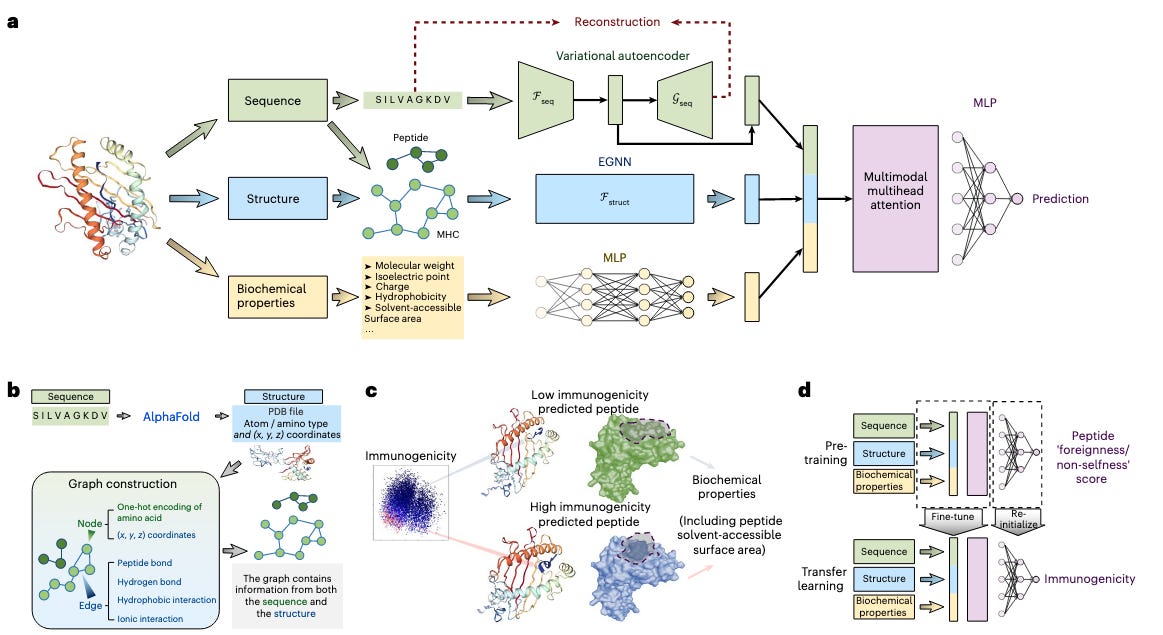
Unlike its predecessors that model peptide-MHC interactions using only sequence data as input, ImmunoStruct also includes structural and biochemical information in its framework and uses a range of architecture choices to process them accordingly. First, the model uses a variational autoencoder to compress peptide-MHC sequences into a continuous latent space to learn essential sequence motifs. In parallel, an equivariant graph neural network processes 3D structures of the peptide-MHC complexes generated by AlphaFold2 to capture spatial relationships and atomic interactions that dictate peptide-MHC binding. Finally, properties like hydrophobicity and solve-accessible surface area are encoded with a multilayer perceptron to generate an embedding of biochemical features. These three representations are fed through a multimodal attention mechanism and final MLP module to predict a simple immunogenicity score. Crucially, the attention mechanism is used in downstream analysis to search for interpretable features that the model has learned during training. ImmunoStruct was trained on a bespoke curated dataset of approximately twenty six thousand peptide-MHC sequences from the Immune Epitope Database (IEDB) and Cancer Epitope Database and Analysis Resource (CEDAR) that included infectious disease and cancer neoepitopes respectively.
ImmunoStruct demonstrated strong performance on held-out data from IEDB, especially in comparison to prior tools that only use sequence information, with an AUPRC of nearly 0.7 in contrast to only 0.46 from the next most performative model BigMHC-IM. Additionally, the authors also tested if ImmunoStruct was robust against genetic diversity and excluded the ten rarest MHC alleles from training; in spite of this, the model was able to maintain strong predictive performance. The team also extended ImmunoStruct to predicting immunogenicity of SARS-CoV-2 epitopes that were validated through in vitro ELISpot interferon-γ assays. Furthermore, the model was also able to successfully predict differential immunogenicity between cancer isoforms of KRAS-GV12 and G12D, crucially being able to match known clinical observations where the latter had a stronger T cell response. Finally, the authors tested ImmunoStruct at the level of patient cohorts, evaluating if the model could stratify patients (who were undergoing various immunotherapies) into high and low risk groups based on a predicted neoantigen load. For each patient, the authors identified all unique peptide-MHC pairs and used ImmunoStruct to score immunogenicity for each pair. Using tumor mutational burden (TMB) as a benchmark, the model was found to be a significantly more accurate predictor of patient survival.
In summary, ImmunoStruct demonstrates the potential of incorporating additional data modalities like structure and biochemical properties to augment the abilities of sequence-only models. While clearly a positive step towards improved vaccine design capabilities, the authors also deploy a host of interesting modeling techniques like multiheaded self-attention and contrastive loss while embedding wild type and mutated sequences to improve interpretability and overall performance.
A multimodal sleep foundation model for disease prediction [Thapa et al., Nature Medicine, January 2026]
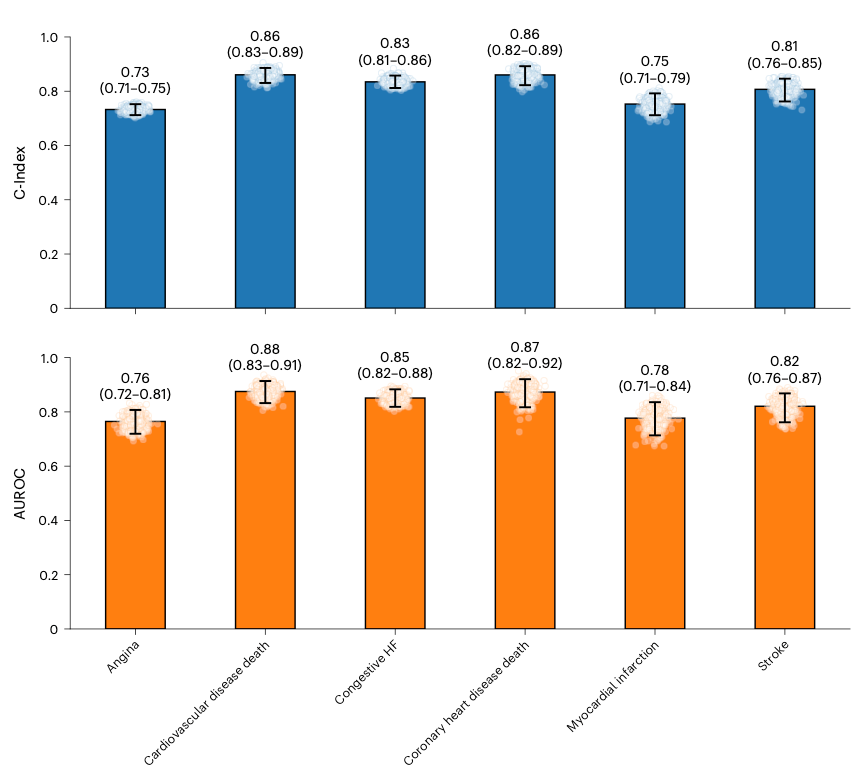
Why it matters: the authors reported that a single night of polysomnography (PSG) contains enough information to predict a large number of diseases, with 130 reaching high prognostic accuracy. The model achieved an AUROC of above 0.75 for various indications (as seen above), supporting that sleep could be a systemic biomarker. This could be used for risk stratification, decision support and triage. This could also be transferred to wearables in the future for large-scale, non-invasive monitoring.
SleepFM is a large sleep foundation model that learns physiological representations from PSG, predicting a wide range of possible diseases from a single night’s sleep, outperforming conventional models.
SleepFM was trained on over 585,000 hours of PSG from 65,000 participants across several cohorts using EEG, EOG, ECG, EMG and respiratory signals. These were recorded in a channel-agnostic architecture, meaning its less sensitive to the number and position of channels. From one overnight study, the model achieves high performance on standard sleep tasks (sleep apnea severity and presence accuracy) and predicts 130 future conditions (including CV death, angina, myocardial infarction, heart failure, CKD, stroke, atrial fibrillation).
SleepFM is composed of a CNN and a transformer encoder. The “channel‑agnostic pooling and leave‑one‑out contrastive learning” aligns modalities and is therefore robust to missing or insensitive to position of channels (e.g. electrodes) across sites.
Notable deals
Soley Therapeutics raises $200M in an oversubscribed Series C round led by Surveyor Capital. Funding will be used to support IND-enabling work and clinical trials for Soley’s two lead oncology assets which target acute myeloid leukemia and solid tumours. Their leukemia asset is on track to receive IND approval within the year. In addition to facilitating their clinical efforts, the raise will also be employed to support further development of the company’s proprietary cell stress platform, which utilizes computer vision and AI to monitor and collect cellular responses from the interaction of diverse human cell types with hundreds of thousands of compounds each week. Those data are extrapolated into actionable insights that can be employed to accelerate hit finding, mechanistic insights, and molecule design. Other investors in the round include HRTG Partners and RWN Management which joined as new investors with participation from existing investors Doug Leone Family Fund, Breyer Capital, and GordonMD Global Investments LP, among others.
Rakuten Medical raises $100M in an oversubscribed Series F round led by TaiAx to advance U.S. regulatory approval for their lead photoimmunotherapy asset. This asset, dubbed ASP-1929, is soon to enter global Phase III trials for recurrent head and neck cancers as a combination therapy with pembrolizumab. In the U.S., ASP-1929 is currently in Phase II trials, also for head and neck cancers. The drug is the first to come off of Rakuten’s Alluminox® Platform, which employs photoimmunotherapy technology developed by Dr. Hisataka Kobayashi and team at the National Cancer Institute. These photoimmunotherapies utilize antibodies conjugated to a light-activated dye for specific targeting of cancerous cells and subsequent activation and via local light stimulation. Rakuten’s Series F raise comprised $70M in new funding alongside $30M from convertible promissory notes. Notably, the round included the greatest number of institutional investors in company history, drawing global representation across key markets of the U.S., Japan, and Taiwan.
Mediar Therapeutics raises $76M in an oversubscribed Series B round co-led by Amplitude Ventures and ICG. The company, which focuses on developing drugs to address fibrosis, will use the funds to support two of their assets through clinical trials. MTX-474 is an IgG1 antibody currently in Phase IIa for systemic sclerosis. The drug is designed to halt the EphrinB2 signaling, which is critical to the onset and progression of the disease. MTX-439 is also an IgG1 antibody, engineered to inhibit SMOC2, a protein heavily implicated in renal fibrosis. MTX-439 is soon to enter Phase I for chronic kidney disease. About a year ago, Lilly agreed to license Mediar’s lead asset, MTX-463, for $99M upfront, with the potential for an additional $687M in milestones as the asset moves through and beyond Phase II trials for idiopathic pulmonary fibrosis.
Following the release of Novo’s oral GLP-1, Eli Lilly strikes a second partnership with Nimbus Therapeutics, totalling up to $1.3B for both oral obesity and metabolic targets. The deal constitutes $55M in upfront and near-term milestone payments to Nimbus, with a potential total of the aforementioned $1.3B in endgame development, commercialization, and sales milestones in addition to tiered royalties should the drug eventually go to market. This is the second deal Lilly has done with Nimbus, the first in 2022 being worth up to $496M for metabolic diseases. In addition to their now two deals with Lilly and their own internal programs, Nimbus has also sold two of the assets they have developed using their computational chemistry and structure-based approach, one to Gilead in 2016 for liver diseases and another to Takeda in 2022 for immune-mediated diseases.
GSK has signed a five‑year, non‑exclusive oncology deal with Noetik under which it will pay $50M in upfront and short‑term milestone payments to license Noetik’s AI cell models for non‑small cell lung cancer and colorectal cancer, using them across GSK’s existing pipeline and earlier‑stage research to select likely responders, discover biomarkers, and complement translational work such as patient‑derived organoids.

In case you missed it
HSBC unveils 2025 Life Sciences and Healthcare Venture Report
Mitochondrial transfer from glia to neurons protects against peripheral neuropathy
Polyphron emerges from stealth, announcing their mission of building a scalable, AI-driven tissue foundry to solve chronic disease at its root cause, tissue failure. Backed by Compound, Humba, Atypical, and Boost VC, the company plans to use machine learning, automation, and developmental atlases to repeatedly design and manufacture functional human tissue of clinically relevant size and complexity in pursuit of this goal. Polyphron further explains their method to addressing tissue failure by delivering immune-compatible replacement constructs that restore architecture and function, not just modulate pathways. Read more here.
What we liked on socials channels



Field Trip
Did we miss anything? Would you like to contribute to Decoding Bio by writing a guest post? Drop us a note here or chat with us on Twitter: @decodingbio.
The SPARK-seq approach is clever. Using pooled CRISPR knockouts to map aptamer binding partners sidesteps the traditional SELEX limitation of getting binders without target identification. The dissociation kinetics coupling is especially valuable for therapeutics. I worked on something tangentially related a while back, but without single-cell seq integration the throughput wasnt there. Combining knockout perturbations with binding readouts in parallel is exactly hte kind of dataset ML models need to go from selection to rational design.