

BioByte 136: Scaling Laws for pLMs, DeepSomatic and CASTLE for Oncological Variant Calling, Ballistic Microscopy for Cellular Imaging, Neural Circuitry for Encoding of Inferred Contours




Welcome to Decoding Bio’s BioByte: each week our writing collective highlight notable news—from the latest scientific papers to the latest funding rounds—and everything in between. All in one place.


What we read
Blogs
Do Scaling Laws Hold for Protein Language Models? [Align Bio, October 2025]
An understanding of scaling laws has allowed natural language processing (NLP) researchers to accurately predict the impact of compute, parameters, and data on model loss. Given the relationship, a desired loss target can be selected and the necessary resources to meet that requirement can be derived. With the stagnation of protein variant effect prediction even with ever growing databases, Align Bio sought to answer whether this same sort of empirical relationship can be applied to protein language models (pLMs).
To investigate this question, the team elected to train the AMPLIFY suite of pLMs with a zero-shot approach on yearly snapshots of UniRef100 from 2011 to 2024, holding all else constant. From their results, they found that the increase in performance, as measured by the Spearman correlation, fluctuated quite significantly, and even decreased some years with the addition of billions of new protein sequences. A follow-up experiment which employed a simple linear regression approach with AMPLIFY embeddings did show consistent improvement as more labeled data was added, but a further trial with a targeted β-lactamase dataset exhibited no improvement over the years, implying that the relationship with labeled data is rather nuanced.

From their observations, the authors conclude that the size of the dataset is not the predominant driver of predictive capability, but rather the diversity within the training set is. The later years were often dominated by redundant sequences, and relative composition of the dataset was also in constant flux with events like the COVID-19 pandemic increasing the relative percentage of a specific group of proteins. To address the issues facing reliable scaling laws in biology, the authors emphasize the need for proper data hygiene and dataset diversity, while also highlighting concerns regarding the scarcity of labels and the generalizability of models.
Papers
Accurate somatic small variant discovery for multiple sequencing technologies with DeepSomatic [Park and Cook et al., Nature Biotechnology, October 2025]
Special thanks to the team at Google for reaching out with the paper pre-release!
Why it matters: Accurately classifying the thousands of somatic variants that contribute to cancer is a challenging task in precision oncology research. While the vast majority of previous variant caller tools have focused on using short-read sequencing data, there has been comparatively less development of tools that can leverage long-read sequencing. In this paper, the authors describe an improved long-read variant caller model, DeepSomatic and release the accompanying high quality somatic variation dataset, CASTLE. Apart from showing improved performance over existing variant callers, DeepSomatic shows significant performance gains on real patient-derived samples, demonstrating its potential for further development towards clinical applications.
A key challenge in precision oncology is the accurate classification of somatic mutations in cancer. Crucially, cancers are defined not by a single mutation, but rather a landscape of thousands of mutations across diverse cell populations. Additionally, mutations can either be acquired at birth (germline mutations) or through external factors like radiation and carcinogen exposure (somatic). With somatic mutations playing a greater role in overall cancer progression and development, there has been increasing attention on studying these variants and their consequent effects. The expansion of various sequencing technologies has enabled scientists to measure these myriad mutations; however, accurately identifying and classifying them remains a significant challenge. Short-read sequencing platforms struggle to map highly repetitive regions of the genome, whereas their long-read counterparts are better suited to identifying rare or adjacent mutations at megabase scales. Nonetheless, the increasing wealth of available sequencing data has enabled the development of computational statistical and machine learning tools for variant calling. However, most methods have been built using short-read data, with only a few being tailored for long-reads or both. Additionally, there is a significant shortage in benchmark data for somatic variant detection, especially when compared to the wealth of information available for germline mutations. To address these problems, the authors present the state-of-the-art DeepSomatic variant caller model and Cancer Standards Longread Evaluation (CASTLE) dataset.
DeepSomatic adapts the DeepVariant model from 2018 meant for germline variant classification, specifically incorporating tumor and germline variation information into the model’s convolutional neural network stages to make it more suitable for somatic mutation classification. Broadly speaking, the model uses a three-step process to classify somatic variants from tumor-normal data. First, the model curates a set of candidate variants from normal and tumor sample reads. Next, the reads from which these candidates originate are then converted into tensor representations that incorporate additional information about overall read and mapping quality. Finally, the model’s convolutional layers classify the candidates as normal, sequencing errors, germline variants, or somatic variants.
The authors first trained single-cancer versions of DeepSomatic on the Sequencing Quality Control 2 (SEQC2) project, the only publicly available high quality benchmark for somatic variants, albeit with only approximately forty thousand mutations from a single cancer cell line. While each version was trained on different long-read platform data, the overall generalizability of these models was limited by the overall training data scarcity and diversity. To mitigate this problem, the authors performed long-read sequencing on four breast cancer and two lung cancer cell lines, obtaining Oxford Nanopore, PacBio HiFi, and Illumina sequencing data to augment the SEQC2 dataset. To actually classify the variants in this new data, the single-cancer models were used to curate high confidence predictions. Specifically, a variant was classified as somatic only if two of the three single-cancer models designated it as such. This process yielded the CASTLE dataset, with the long-read sequencing data and variant predictions made publicly available.
Armed with the CASTLE dataset, the authors then trained multi-cancer versions of DeepSomatic, split by sequencing platform (Nanopore, HiFi, or Illumina). DeepSomatic multi-cancer models showed superior performance on an orthogonal technology benchmark, which measured a model’s overall consensus with existing methods. Notably, the model showed a substantial decrease in error rate of 95% when classifying indel variants and 37% when classifying single nucleotide variants. Crucially, DeepSomatic also showed high accuracy when evaluated on held-out cell lines, demonstrating more robust generalizability. Furthermore, the model also excelled at low variant allele frequency tasks, achieving the highest recall values for variants with less than 10% allele fraction. Finally, DeepSomatic was also applied to real patient data, specifically glioblastoma and pediatric blood cancers, to prove its capabilities on samples with higher subclonality. The model demonstrated higher recall performance on the glioblastoma sample when compared to the ClairS method, and was also able to identify ten additional somatic mutations not found by ClairS in pediatric blood cancers, although it did miss two other mutations.
DeepSomatic and CASTLE are significant steps forward in variant caller capability, with perhaps the biggest contribution being the massive jump in publicly available sequencing data and high confidence variant prediction. In terms of future improvements, the authors point to the inclusion of additional open source sequencing data to better capture heterogeneous mutational signatures and overcome low sequencing depth limitations. It will be interesting to see how the improved variant calling abilities enhance precision oncology research, potentially by allowing clinicians to better characterize complex tumors and detect subtle, low-frequency subclonal mutations in patient samples.
Ballistic Microscopy (BaM) [Jijumon et al., bioRxiv, October 2025]
Why it matters: Studying single cells with spatial context is really hard. Methods either sacrifice molecular characterization for great spatial resolution (like CryoET), or rely on tagging known spatial proteins to anchor studies in specific parts of the cell. Jijumon et al. decouple spatial extraction and molecular by introducing Ballistic Microscopy (BaM), where high-speed nanoparticles are blasted through living cells to extract tiny attoliter droplets of cytoplasm onto hydrogels, preserving the cell’s spatial information for later molecular analysis, akin to taking a molecular photograph onto a hydrogel film. This unlocks unlabeled exploration of spatial niches inside living cells, and can possibly even be extended to full tissue samples.
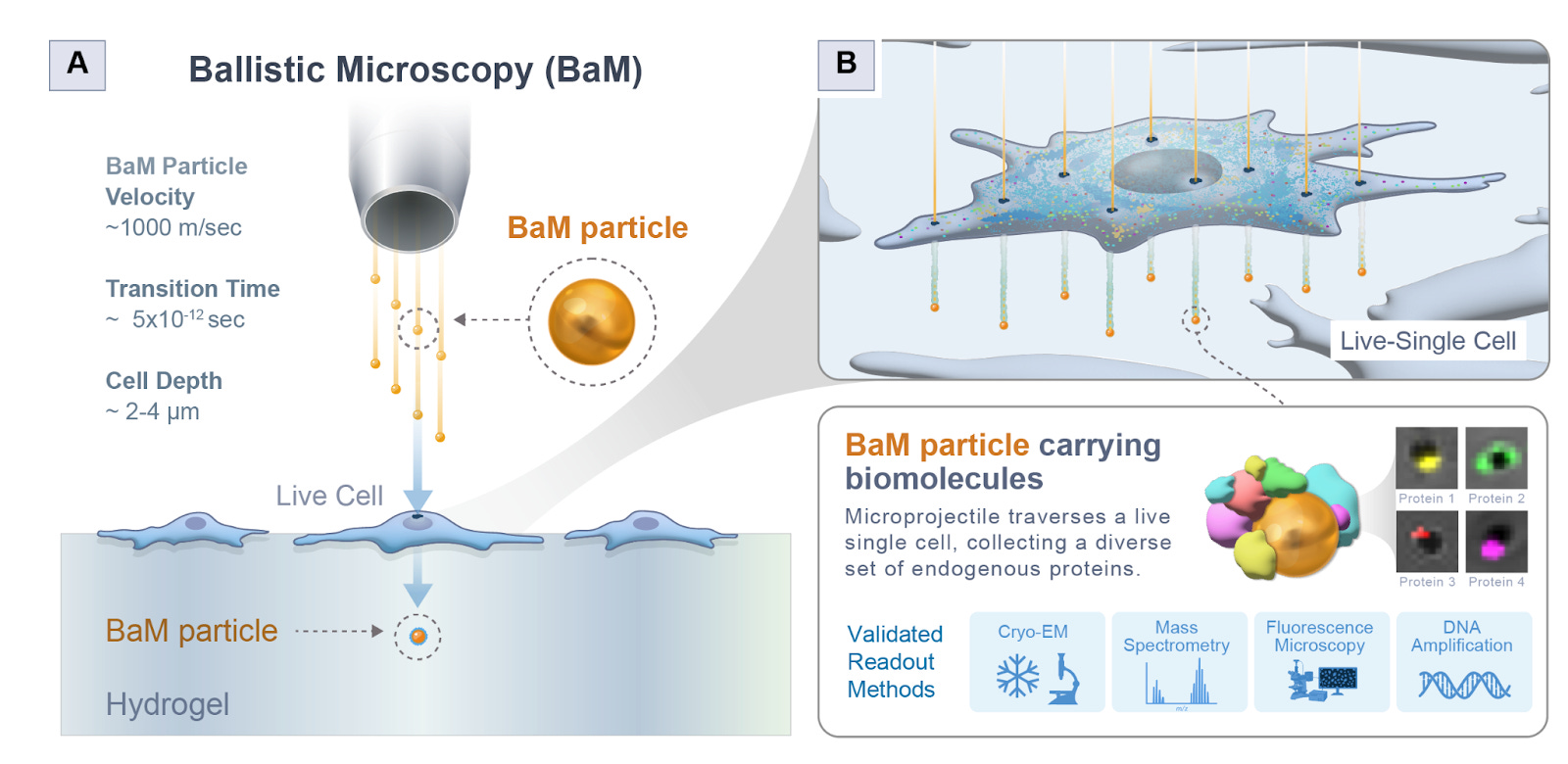
They first checked theoretically if this could even work. Essentially, BaM fires gold nanoparticles at around 1000 m/s - fast enough to fast enough to pierce a cell but not so fast as to vaporize or destroy it. Each particle passes through the cell, dragging a femtoliter-scale droplet of cytoplasm that then lands on a hydrogel beneath, forming what they call a SPLAT-MAP—a physical image of the cell’s contents. To ensure it was physically feasible, they modeled heat transfer, cavitation, drag, and penetration depth. They showed that the particles wouldn’t overheat (transient temperature rises only ~200K for a microsecond), would not cavitate beyond 0.2 μm, and could safely traverse the cell membrane without blowing it apart. They also estimated that each nanoparticle could collect around 20 attoliters of cytoplasm, containing on the order of 105 proteins and up to 5 RNA molecules, enough for high-content molecular assays.
First, they tested BaM in a controlled system by coating gels with GFP-actin lysate and firing particles through it. The particles picked up GFP signal, confirming protein transfer. Then they tested whether nucleic acids could be captured intact: when they blasted particles through a gel with LAMP reagents, those same particles could still amplify DNA, showing that the process doesn’t destroy molecular material. They then moved to live HEK293 cells, showing through confocal imaging that BaM particles collected nuclear and membrane material in a spatially consistent pattern. Finally, Cryo-EM confirmed that BaM particles carried intact, membrane-enclosed vesicles of cytoplasm on their surfaces.
They used BaM to study condensates formed by the cytoskeletal protein CLIP170. Because each particle captures attoliters of material, they pooled thousands of GFP-tagged CLIP170 particles and ran mass spectrometry. The proteomic analysis revealed 641 associated proteins forming a dense interaction network. Among them, Keratin-18 emerged as a consistent and surprising interactor. Follow-up confocal imaging confirmed Keratin-18 co-localized within CLIP170 condensates, and similarly, Tau3R condensates were found surrounded by cage-like shells of Keratin-18 - a discovery that points to intermediate filaments as key structural organizers within condensate architectures.
BaM separates capture from analysis - like taking a physical film photograph of a cell that you can later develop through mass spec, cryo-EM, or sequencing. This modularity lets you combine destructive techniques on the same spatial sample. They note current limitations: attoliter sampling means very small material quantities, requiring pooling or ultrasensitive MS. There’s no z-depth resolution yet, and mechanical shock from gas acceleration can limit repeated sampling. Future versions could use laser-based projectiles or targeted nanoparticle coatings to probe specific organelles. But overall, this is an insanely creative piece of engineering - shooting little bullets to form a splatter map of a cell is a wild idea that hopefully will never be accepted as obvious! The authors attached some capture videos in their supplementary material, which the reader is recommended to check out.
Recurrent pattern completion drives the neocortical representation of sensory inference [Shin et al., Nature Neuroscience, September 2025]
Why it matters: Perception routinely requires the brain to infer whole objects from partial, ambiguous inputs. Understanding which cells and circuits in the cortex construct those inferences links cellular physiology to core computations of vision. Shin et al. provide that link by identifying a sparse population in the primary visual cortex that encodes inferred contours and demonstrating that those neurons can causally recreate the inferred percept through recurrent local pattern completion. More broadly, this finding may point to a compact, generalizable circuit motif – a mechanistic template that could inform resource-efficient models of perception in biology and artificial systems.

Illusory contours (IC) – like the Kanisza triangle, where viewers perceive edges that aren’t physically present – offer a tool for dissecting how brains construct perception from incomplete information. Prior work showed that neurons in the primary visual cortex (V1) respond to such illusions despite no contrast in their receptive fields, and that silencing visual areas reduces these responses – implying top-down feedback. However, whether feedback signals merely modulate activity or play an active, causal role remained unclear.
Using large-scale Neuropixels recordings and two-photon calcium imaging across the mouse visual cortex, the authors discovered that a small fraction (~4 – 8%) of layer 2/3 V1 neurons responds selectively to illusory bars but not to the inducing segments. Population decoders trained on L2/3 activity reliably discriminated IC stimuli from controls and generalized to classify real edges, indicating that the L2/3 population carries a robust, emergent representation of the inferred contour. Cross-area analyses further showed that these IC-encoders receive stronger putative excitatory input from higher visual areas than do the segment-responsive cells, consistent with top-down predictive input converging on a sparse V1 subpopulation.
To test whether the IC-encoders causally drive the representation of these inferred contours, the team implemented a closed-loop optical pipeline: imaging thousands of neurons, identifying functional ensembles in real time, and selectively stimulating them with two-photon holographic optogenetics. Activation of IC-encoder ensembles (15-18 cells on average) in the absence of visual input recreated the neural pattern normally elicited by the illusory contour across the remaining V1 L2/3 network. Decoders trained only on visual responses classified this stimulated activity as the corresponding illusory stimulus with 40% accuracy (chance = 25%) – approaching the 48% cross-validation accuracy on held-out visual trials. By contrast, stimulating segment responders failed to drive the same local inference pattern and instead propagated signals to higher visual areas. Mesoscope experiments (large-field imaging) spanning multiple visual areas confirmed this division: segment responders drive feedforward communication while IC-encoders implement local pattern completion within V1.
The results support a circuit motif for sensory inference in which higher areas broadcast predictions to a sparse set of L2/3 neurons that locally and recurrently amplifies inference-consistent activity to pattern completion, while feedforward segment responders convey sensory evidence upward (bottom-up). The study’s limitations include the small IC-encoder population, experimental setups relying on head-fixed mice with controlled stimuli, and calcium imaging’s dynamics operating on slower timescales than spiking. Subsequently, establishing behavioral relevance, testing generalization to naturalistic occlusion, mapping the synaptic connectivity underlying IC-encoders privileged top-down access, and determining whether similar motifs support other inference problems are clear directions that would cement this mechanistic bridge between circuit physiology and perceptual inference.
Notable deals
Kailera Therapeutics announced a $600M Series B round led by Bain Capital Private Equity. This news follows the company’s impressive exit from stealth with a sizable $400M Series A just last year. The funding will advance Kailera’s substantial obesity portfolio, including leading candidate, KAI-9531, an injectable GLP-1/GIP receptor agonist that is entering Phase III trials and is striving for best-in-class designation—potentially posing fierce competition for Eli Lilly’s Zepbound. Also in the portfolio is KAI-7535, an oral small molecule GLP-1 receptor agonist that has shown very promising results in Phase II clinical trials in China. The exclusive rights to this myriad of strong obesity assets as well as several other metabolic disease indications was acquired by Kailera from Jiangsu Hengrui Pharmaceuticals for global development outside of greater China in May 2024. Kailera’s projected success and the profitability surrounding the GLP-1 craze makes the company a strong contender for future biopharma acquisitions. Other investors in the round include: Adage Capital Management LP, CPP Investments, Invus, Janus Henderson Investors, Perseverance Capital, Qatar Investment Authority, Royalty Pharma, Surveyor Capital, accounts advised by T. Rowe Price Associates, Inc., and an undisclosed large mutual fund, as well as existing investors: Atlas Venture, Bain Capital Life Sciences, RTW Investments, and Sirona Capital.
Only a month following an initial $235M Series A close, Lila Sciences disclosed an additional $115M in funding as a Series A extension. The mission of building what the company terms “scientific superintelligence” remains the same, however, the aim is now further bolstered by the support of NVentures, Analog Devices (part of existing partnership with Flagship), IQT, Dauntless Ventures, Catalonia Capital Management, Pennant Investors, and a group of investors from the Peter Diamandis’ Abundance Membership, amongst other new stakeholders. This extension brings Lila’s Series A to an admirable $350M, and the company’s total capital raised to $550M.
RADiCAIT emerged from stealth with a $1.7M preseed round led by Frontline Ventures. The company is seeking to revolutionize the medical imaging and diagnostic space using a novel AI foundational model to create synthetic PET images from CT scans. Spun out from Oxford, RADiCAIT’s mission is to drastically increase safety, accessibility, and productivity of the existing technology’s diagnostic capabilities by reducing the reliance on nuclear medical facilities that are far more costly and pose significant risks to patients due to usage of radioactive tracers. Still early in development, the tech is still awaiting FDA approval. Other investors in the round include Gurtin Ventures, healthcare industry angels, and Techstars.
Kardigan Therapeutics raised a $254M Series B to advance their cardiovascular- focused portfolio. The company seeks to develop its three lead assets in tandem, each addressing one of acute severe hypertension (ASH), specific types of dilated cardiomyopathy (DCM), and calcific aortic valve stenosis (CAVS), with the hope that these drugs will be able to treat the root cause of the diseases rather than simply manage the symptoms. This current fundraising round follows their $300M Series A in January of this year, with participants in the Series B including new investor Fidelity Management & Research Company and continued support from existing investors ARCH Venture Partners and Sequoia Heritage.
In a round led by Engine Ventures, Bexorg raised a $23M Series A for their integrated AI and whole-human brain platform. Founded out of Yale in 2021, Bexorg is working to address the 95% failure rate of CNS drugs in clinical trials. Utilizing both diseased and non-diseased donated human brains, the company is able to produce datasets that afford a better understanding of a molecule’s interaction with human biology, data which is subsequently fed into AI models to accelerate and improve the decision making process. The proceeds from the round will go towards expanding Bexorg’s AI platform and the advancement of their programs. New investors in the round included Connecticut Innovations and E1 Ventures, with additional support from existing investors Amplify Partners and Starbloom Capital.
Takeda entered into a second research partnership with Nabla Bio for use of Nabla’s proprietary AI drug discovery platform. This multi-year agreement represents an expansion of the existing partnership between the two companies, and emphasizes big pharma’s continued—and deepening—interest in AI for drug development and discovery. In the partnership, Nabla’s platform, Joint Atomic Model (JAM), which CEO Surge Biswas describes in coverage by Reuters as functioning like ChatGPT in that it “responds to molecular queries by designing antibodies from scratch that bind targets with desired properties,” will design protein-based therapeutics for Takeda’s early-stage pipeline, focusing on hard-to-treat diseases. The deal confers an undisclosed double-digit million upfront value to Nabla as well as up to $1B in success-based milestone payments. Nabla Bio boasts a timeline of 3-4 weeks from design to lab testing for their platform, claiming it as “probably the fastest feedback loop in the industry,” and anticipates first-in-human data from their AI-designed candidates within a mere 1-2 years.

What we liked on socials channels



Field Trip
Did we miss anything? Would you like to contribute to Decoding Bio by writing a guest post? Drop us a note here or chat with us on Twitter: @decodingbio.
 |
|