

BioByte 135: improvements to biosecurity screening, CPTAC-PROTSTRUCT and KRONOS demonstrate LLM fine-tuning for patient-specific prognoses, new methods for imaging at single-protein resolution




Welcome to Decoding Bio’s BioByte: each week our writing collective highlight notable news—from the latest scientific papers to the latest funding rounds—and everything in between. All in one place.


What we read
Papers
Strengthening nucleic acid biosecurity screening against generative protein design tools [Wittman et al., Science, October 2025]
Why it matters: Nucleic acid screening companies are the watchtowers that ensure that new strands of DNA being ordered are not harmful for humanity. As deep learning has transformed protein design, bad actors’ ability to design harmful sequences becomes more robust. Researchers from Microsoft led a red-teaming collaboration to help nucleic acid screening companies improve their ability to catch potentially toxic proteins that look benign in sequence space but could be troublesome based on their structure.
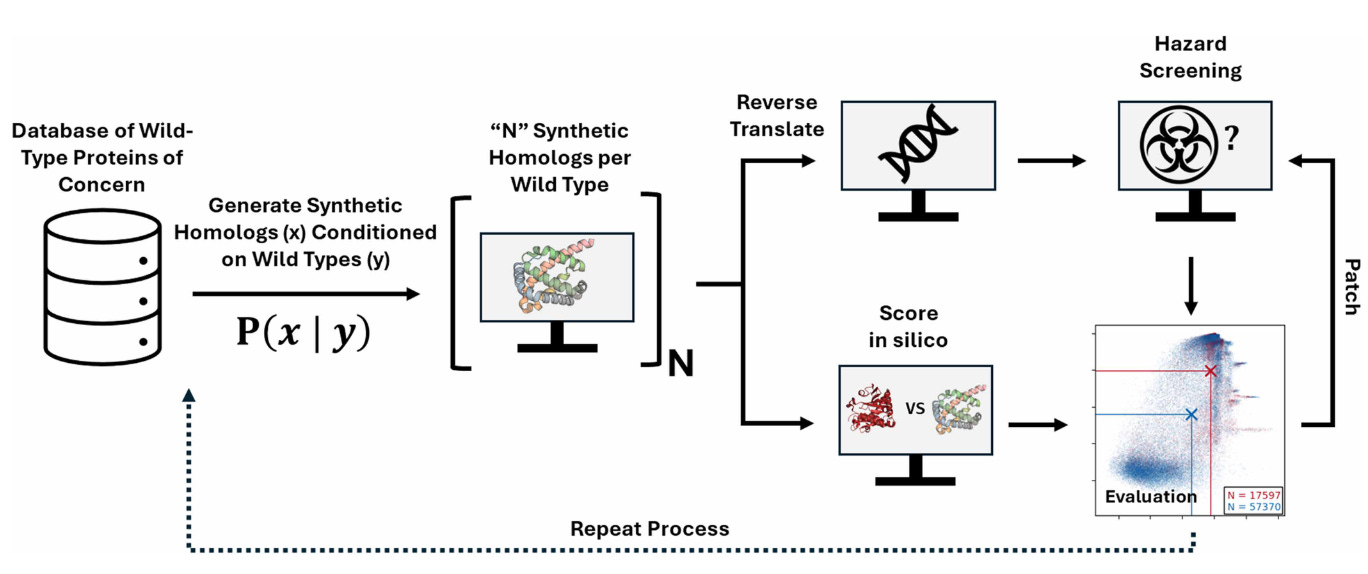
Historically, biosecurity screening has relied on sequence-based homology searches. Methods like BLAST enable scientists to flag sequences if they closely resemble a known toxin or pathogen gene. This approach worked because dangerous proteins were generally sequence-similar to previously cataloged ones. But with modern deep-learning tools, we can now predict 3D protein structures with remarkable accuracy and compare them using structure-based tools like FoldSeek, revealing similarities that sequence alone might miss. At the same time, generative protein-design models such as ProteinMPNN and EvoDiff can create entirely new sequences that differ dramatically in amino-acid identity yet still fold into the same functional structures as known toxins. To test and strengthen global biosecurity infrastructure, Microsoft researchers conducted a biosecurity red-team exercise with synthesis providers to “attack” and stress-test their screening systems using AI-designed proteins meant to evade detection.
The team computationally generated 76,080 synthetic homologs derived from 72 wild-type proteins of concern (including ricin, botulinum neurotoxins, and viral effectors). Each variant was designed using open-source generative models at different mutation levels to test how far sequence drift could go while preserving structure. Although the study did not synthesize or experimentally validate the proteins, structure predictions with OpenFold showed that many variants retained folds strongly resembling the originals - meaning they should, in principle, trigger screening alerts. The researchers also explored a DNA-obfuscation strategy, in which protein-coding sequences were fragmented, scrambled, and reversed across reading frames to mimic how a malicious actor might further disguise intent from DNA-level screens.
The red-team exercise revealed that existing “best-match” sequence-based frameworks missed a substantial fraction of these AI-reformulated toxins. In response, three major biosecurity-screening providers (Aclid, IBBIS, and RTX BBN) rapidly updated their pipelines, incorporating profile HMMs, regex-generalized k-mer signatures, and machine-learned functional annotations. These “patched” systems could now flag ≈97 % of likely functional synthetic homologs, up from near-zero detection before. Tests of the DNA-obfuscation attack further showed that fragment length and reading-frame scrambling can hide sequences from naive screens—highlighting that next-generation biosecurity must combine sequence, structure, and contextual threat modeling to remain effective in the age of AI-driven protein design.
Patient-specific Biomolecular Instruction Tuning of Graph-LLMs [Adam et al., arXiv, September 2025]
Why it matters: While conventional cancer research has placed heavy emphasis on molecular genomics and mutational signatures, the availability of additional data forms like high-throughput proteomics have given researchers more real-time snapshots of the disease. However, effectively leveraging this wealth of information to yield useful patient-specific clinical interpretation remains challenging. Although large language models have shown exceptional ability in handling multimodal data (like text and images) and in associated reasoning tasks, current methods are unable to effectively reason over proteomics data and produce “individualized semantic molecular reasoning.” In this paper, a team from the new startup, Standard Model Bio, present CPTAC-PROTSTRUCT and KRONOS. The former is “the first patient-level instruction tuning dataset for molecular oncology understanding,” while KRONOS is a graph-LLM that combines patient-specific proteomics data and more general molecular information to develop individual prognoses. Together, these advancements show a potential way forward to capitalize on the immense power of LLM frameworks and high-throughput patient-specific biological data to guide clinical decision making.
Apart from simply measuring the proteome, graph-based methods have enabled researchers to model diseases by building intricate networks of molecular interactions like protein-protein interactions. To that end, graph neural networks and related techniques have demonstrated the ability to reason over molecular networks for tasks like cancer drive gene identification. On the LLM side, instruction tuning, where a model is finetuned on instruction-response pairs, has helped develop specialized models that can handle a mix of biological and clinical information. However, there are no instruction-response datasets that go between proteomics data for cancer and associated clinical outcomes.
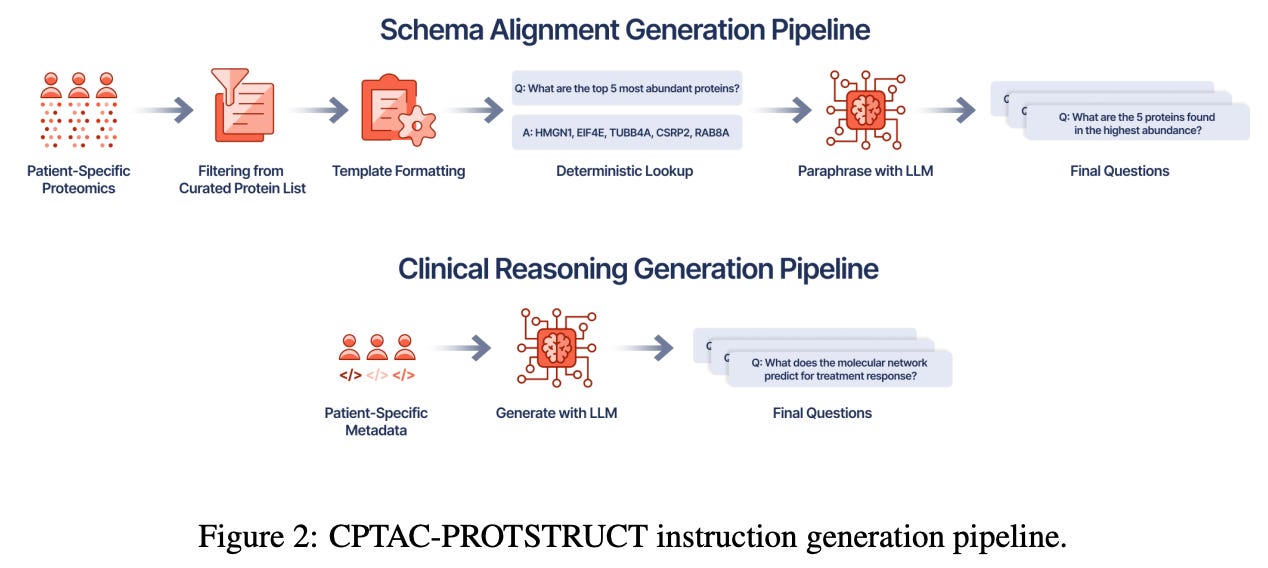
To that end, the authors developed the CPTAC-PROTSTRUCT dataset, using patient data from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) study. After filtering patient samples for significantly sparse protein data, the team identified a set of “core” proteins (to shrink the overall feature space) present in the majority of tumor and normal samples, using imputation methods to fill in any gaps, before finally eliminating some of the lowest variance proteins. On the instruction data side, the team used curated PPI networks to selectively query the processed CPTAC dataset, choosing proteins with high degree centrality within their networks while also relying on pathway analysis to identify proteins that regulated core cellular processes. To incorporate more cancer specific information, the OncoKB and COSMIC database were used to identify cancer-associated proteins that were known drivers of oncogenesis, as well as their known therapeutic response. To actually translate this information into clinical directives, the team used the DeepSeek-R1-Distill-Qwen-32B LLM to “ generate QA pairs that resemble those oncologists might ask when interpreting patient proteomic profiles in clinical settings.”
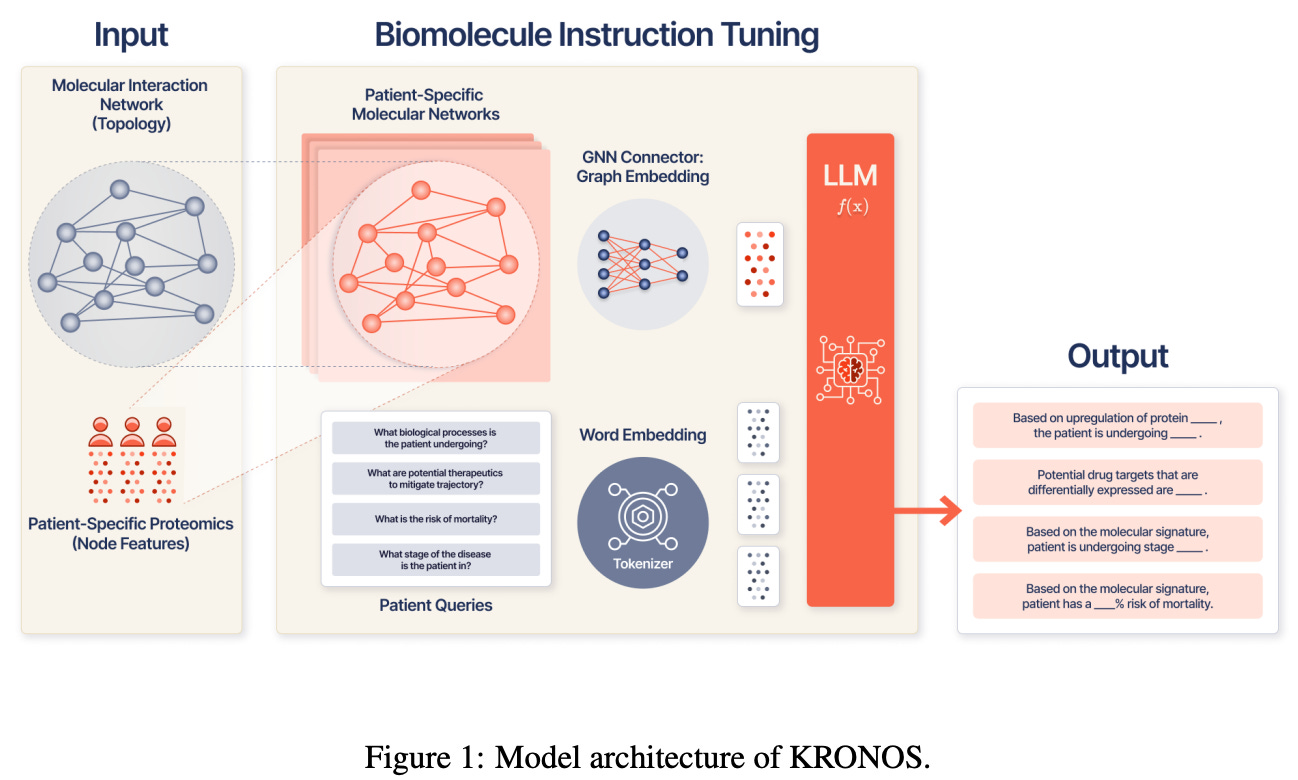
Following up on this dataset, the authors then developed the KRONOS (Knowledge Representation of patient Omics Networks in Oncology via Structured tuning) model. Taking a patient’s proteomic profile as input, KRONOS begins by creating node features for proteins that are then embedded into a broader PPI network graph. Next, a graph neural network module operates over the PPI graph to create a specific representation that is then passed on as a token to a general LLM (in this case, the DeepSeek Qwen model). This overall model was trained in two stages, first to develop a useful representation space that “directly aligns with the semantic space of the LLM,” followed by fine tuning with held-out question-answer pairs to further train the LLM element.
To evaluate KRONOS, the team compared their model against conventional linear modeling approaches, simple multilayer perceptron algorithms, patient similarity network node classification models, and biomolecular graph classification approaches. Specifically, each method was evaluated on mortality prediction, cancer type classification, overall survival estimation, and disease stage prediction. KRONOS achieved the highest performance on all four tasks, with MLP methods also performing competitively. The authors hypothesized that linear methods fail due to their assumptions of independence between protein features, showing the value of explicit PPI network information. In terms of limitations, the team identified future work in proving the generalizability of their method under distribution shifts by evaluating KRONOS on other institutional datasets. Furthermore, the overall computational footprint of the model may pose some challenges in its clinical deployment. Nonetheless, CPTAC-PROTSTRUCT and KRONOS-type approaches demonstrate the potential to develop crucial patient-specific clinical diagnoses based on increasingly complex biological data.
Left-handed DNA for efficient highly multiplexed imaging at single-protein resolution [Unterauer et al., Nature Communications, October 2025]
Why it matters: Understanding how proteins organize at the nanometer scale is central to questions from synaptic function to organelle contacts, yet high-plex, single-protein mapping forces a tradeoff between resolution and throughput. Unterauer et al. break this bind: using left-handed (mirror-image) DNA probes and a streamlined serial-imaging workflow, the authors generate 13-target, 5-nm precision protein atlases across a 200 x 200 micrometer field in 10 hours. This sizable leap in speed and scale unlocks the ability to conduct routine, population-level experiments linking nanoscale architecture to cell state and function.
The core novelty proposed is a clever twist on a super-resolution probing technique called DNA-PAINT. As a brief primer, DNA-PAINT works by using the transient binding of a fluorescently-labeled DNA ‘imager’ strand to a complementary ‘docking’ strand attached to the protein of interest via an antibody; localizing thousands of these binding events builds a super-resolved image. To visualize a wide array of targets, DNA-PAINT is often conducted in rounds, where one imager is washed out before the next is introduced – a method referred to as Exchange-PAINT. However, scaling this process introduces challenges. If imagers have slow (‘sticky’) binding kinetics, erasure chemistries such as toehold displacements are needed. Moreover, to extend the utility of a small set of imager strands to a large set of target proteins, secondary DNA barcodes – or adaptors – are introduced at the expense of increased complexity and noise.
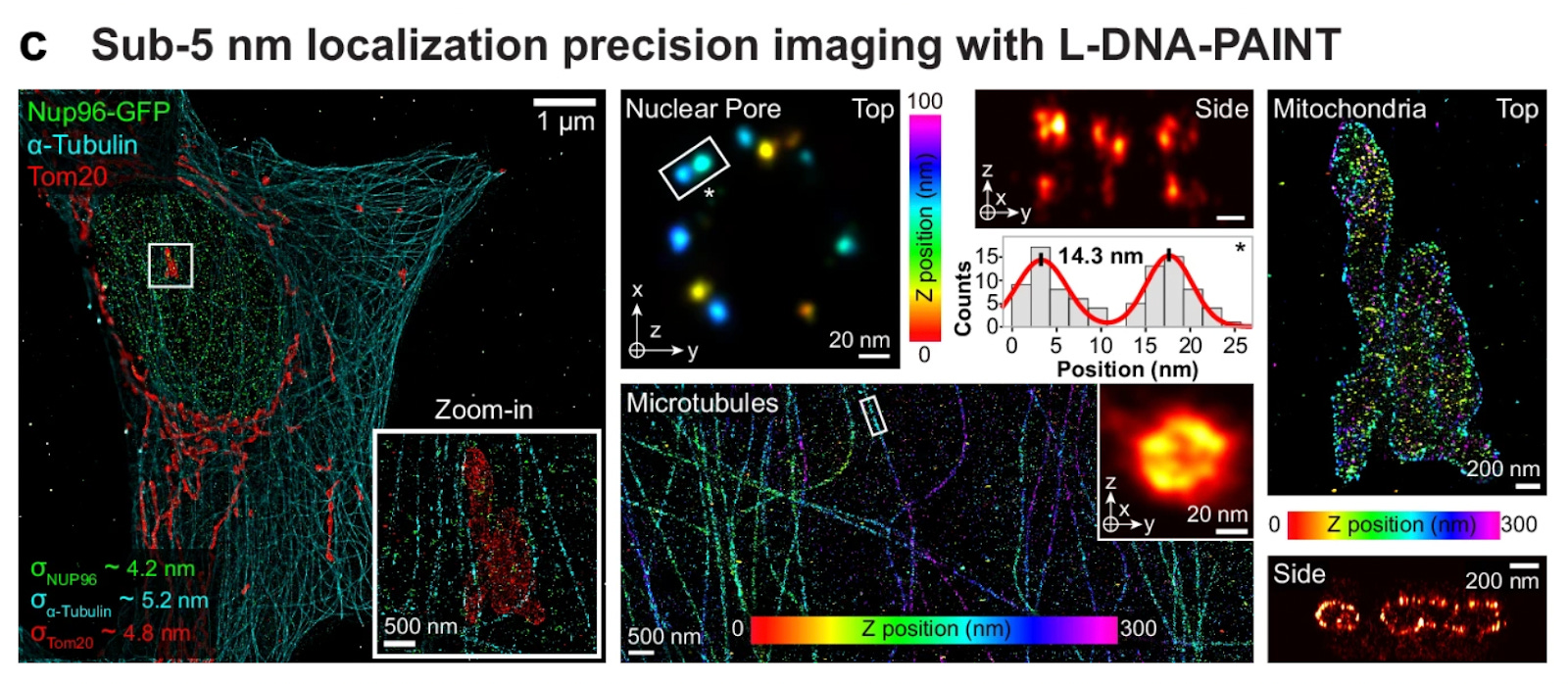
The technical approach exploits an elegant observation: left-handed DNA-PAINT, originally developed to minimize non-specific nuclear binding, represents ‘free’ orthogonal sequence space requiring no new chemistry or optimization. The team designed six left-handed (L) imager strands as hairpin-free, speed-optimized complements to an existing set of six right-handed (R) sequences, validating orthogonality and kinetics on DNA origami (resolving 5-nm features) and cellular benchmarks (Nup96 nuclear pores spaced 14nm apart, Tom20 mitochondrial membrane proteins, ɑ-tubulin filaments). Since L and R strands do not cross-hybridize, the authors could mix both libraries in a straightforward Exchange-PAINT protocol – no secondary DNA barcodes or displacement chemistry needed. This simplification dramatically reduces implementation barriers (e.g. sequence design complexity) while maintaining the speed advantages of optimized binding kinetics in specialized predecessors such as FLASH-PAINT and SUM-PAINT.
The paper is a notable advance, but it’s not yet an ‘out-of-the-box’ solution: L/R probe pairs exhibit small, context-dependent kinetic differences and behaviors that change from DNA-origami to crowded cellular environments, indicating the need for empirical tuning of imager concentration, frame timing, and acquisition parameters. Achieving sub-5 nm localization also depends on upstream factors – validated primary affinity reagents (antibodies / nanobodies), gold-fiducial alignment (using nanoparticles as fixed points to overlay images from all rounds), drift correction, and optimized buffers – that present stronger difficulties in thicker or heterogeneous tissue versus cultured cells. However, the upside remains significant: Unterauer et al. remove notable engineering frictions, effectively double usable channels, and shorten per-target imaging time so single-protein, nanoscale atlases across hundreds of microns move from bespoke, specialist feats toward routine experiments. With systemic mapping of L/R kinetics across biological contexts, broader validation in tissue slices and diverse cell types, and the creation of standardized reagent panels and imaging protocols, this method can democratize high-plex, nanoscale proteomics.
Human gut bacteria bioaccumulate per- and polyfluoroalkyl substances [Lindell., Nature Microbiology, October 2024]
Why it matters: Per- and polyfluoroalkyl substances (PFAS), often referred to as ‘forever chemicals’, are pollutants that pose major environmental and health concerns. This study showed that bacteria, such as Bacteroides uniformis, showed high PFAS accumulation up to a millimolar intracellular concentration while maintaining their viability. If the human microbiome concentrates PFAS and increases fecal elimination, it could change systemic exposure levels.
PFAS is found in water, food and household products.This paper observes how bacteria from the human gut microbiome accumulate PFAS. Bacteroides uniformis concentrated it at high levels, while E. coli accumulated less unless its efflux pump was disabled. Cryogenic FIB-SIMS imaging shows that the bioaccumulated molecules aggregate in dense clusters, which minimizes interference with cellular processes which allows them to survive.
The team also demonstrated that mice colonized with human gut bacteria excreted more PFNA in feces than germ-free controls or those with low-accummulating bacteria, showing that microbiota can sequester PFAS in vivo.
Notable deals
Affinia Therapeutics closed a $40M Series C round led by NEA. A developer of adeno-associated virus (AAV) gene therapies for debilitating cardiovascular and neurological diseases, Affinia plans to use the funding to advance their lead program, AFTX-201, into clinical trials aiming for IND submission in Q4 of this year. AFTX-201 is seeking first-in-class and best-in-class statuses for treatment of BAG3 dilated cardiomyopathy, a serious genetic condition resulting in early onset heart failure with a rapid progression and few current treatment options—factors contributing to the disease’s high mortality rate. The company also boasts a generative AI discovery platform utilized for engineering novel myotropic capsids which specialize in drug delivery to heart and skeletal muscles while avoiding the liver, a common challenge facing gene therapies. Other participating investors included Alexandria Venture Investments, LLC, Atlas Venture, Avidity Partners, F-Prime, GV, Mass General Brigham Ventures, and Perceptive Advisors with Eli Lilly & Company joining the fray as a new investor.
Peer AI raised a $12.1M preseed/seed round led by Flare Capital Partners and SignalFire. Peer is seeking to revolutionize the regulatory documentation space beleaguering the life sciences via domain-specific AI agents. The current FDA approval process requires hundreds of thousands of pages of meticulously specified documentation, severely vulnerable to human error resulting in significant delays, rejections, and thus high capital and time costliness. While clinical trial requirements present a primary use case, the company’s agents also have applications spanning areas such as pre-clinical, CMC, regulatory affairs, and medical writing. In what is attributed to be a $15B market, Peer AI unveils significant and growing adoption of their product—including by biopharma industry leaders—who they claim are now saving thousands of hours across regulatory documentation workflows with substantial quality improvement. Other investors in the financing include Greycroft, Atria, Alumni Ventures, Gaingels, and Mana Ventures, accompanied by several strategic angel investors.
Remedy Robotics secures $35M in funding led by Blackbird Ventures. The round follows their demonstration of the first successful fully-remote endovascular surgery in humans via neurointerventional procedures. The company’s AI-enabled Remedy N1 System allows clinicians to remotely operate on patients from across the globe, conferring potentially life-saving treatment for critical conditions such as heart attack and stroke especially in urgent situations where specialists may not be present. Remedy N1 seamlessly integrates with the cath lab requisite for endovascular procedures, featuring “360-degree catheters for safe navigation through the vasculature, built-in connectivity with image streaming and latency management for seamless remote operation, and an integrated contrast injector that allows operators to manage all aspects of the procedure” as well as live audio-visual communication between the cath lab and remote operator via a portable console, as explained in the official press release. The technology offers unparalleled visibility, precision, and control capabilities and significantly increases accessibility of treatment by virtue of its scalability. Other investors in the round included KdT, DCVC, and Pathbreaker Ventures (among others).
GLD Partners launches Altagenics to develop first-in-class drugs for muscle-wasting indications.The newco will leverage access to Heligenics’ functional genomics GigaAssay platform in the pursuit of discerning novel treatments for sarcopenia and cachexia. While Pfizer is highlighted as also being hot on the trail with a candidate targeting cachexia currently entering Phase III trials, both muscle-wasting disorders still represent a significant unmet need at present. Equipped with GigaAssay, which has capacity for measuring molecular functions of thousands of genetic variants simultaneously, Altagenics and GLD project a multitude of further applications in muscle development and regeneration, sports medicine, rehabilitation, and metabolic health, as well as high scalability of the platform to other therapeutic areas.

What we liked on socials channels



Events

Field Trip
Did we miss anything? Would you like to contribute to Decoding Bio by writing a guest post? Drop us a note here or chat with us on Twitter: @decodingbio.
 |
|